
DRUGONE
研究人员提出了一种名为 ProTrek 的三模态蛋白语言模型,该模型同时整合了蛋白质的序列、结构和功能三种模态。通过对比学习,ProTrek 能在模态之间建立紧密联系,实现跨模态和单模态的多种搜索任务。与现有的序列或结构比对工具相比,ProTrek 在速度和准确性上均表现优异,能够更高效地识别功能相关的蛋白质。计算模拟和实验验证表明,ProTrek 服务器已预先计算了超过 50 亿条蛋白质嵌入,为大规模蛋白质数据库的检索与分析提供了高效平台。

蛋白质是细胞的核心分子机器,驱动着多样的生物学过程。解码序列、结构与功能之间的关系(SSF 关系)是分子科学和药理学的核心挑战。然而,蛋白质的结构多样性、环境依赖的功能特性及复杂的分子互作,使这一任务极具难度。
传统的比对方法(如 BLAST、MMseqs2、Foldseek 等)在推动蛋白研究方面发挥了重要作用,但局限于单模态的两两比对。这种方法不仅限制了跨模态关系的发现,也容易忽视全局上下文,尤其是对于约 30% 未注释的 UniProt 蛋白更为明显。虽然神经网络驱动的工具能够从固定词汇预测功能标签,但无法理解自然语言描述,从而限制了细致功能注释和文本驱动的蛋白检索。
随着大语言模型和蛋白语言模型的快速发展,研究人员提出构建一个基础性模型来统一蛋白的序列、结构和功能模态。
方法
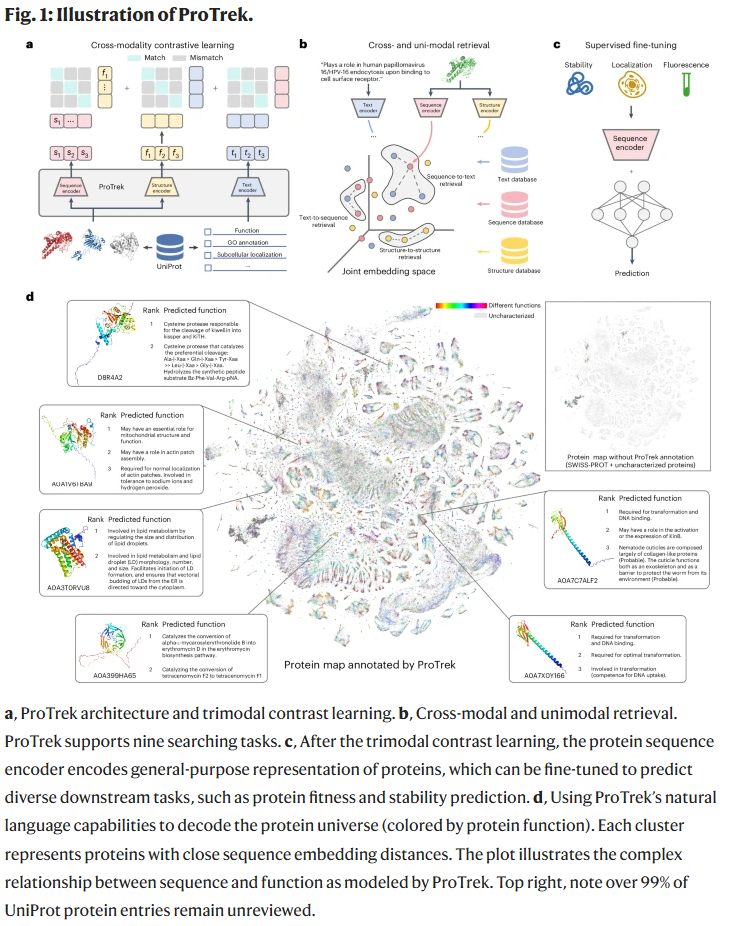
ProTrek 采用三模态对比学习,将真实的序列-结构、结构-功能、序列-功能配对拉近,并将负样本推远。其架构包括:
序列编码器:使用预训练的 ESM 模型。
结构编码器:利用 Foldseek 将蛋白结构离散化为 3Di 序列,再用 BERT 风格模型建模。
文本编码器:采用 PubMedBERT 对自然语言功能描述进行建模。
训练数据包含约 4000 万对蛋白-文本配对,其中包括高质量的 SWISS-PROT 数据和从 TrEMBL 中筛选的“噪声”对。ProTrek 在训练中结合六个跨模态对比损失和两个掩码语言建模损失,从而保证三模态之间的统一表示。
结果
跨模态与单模态检索
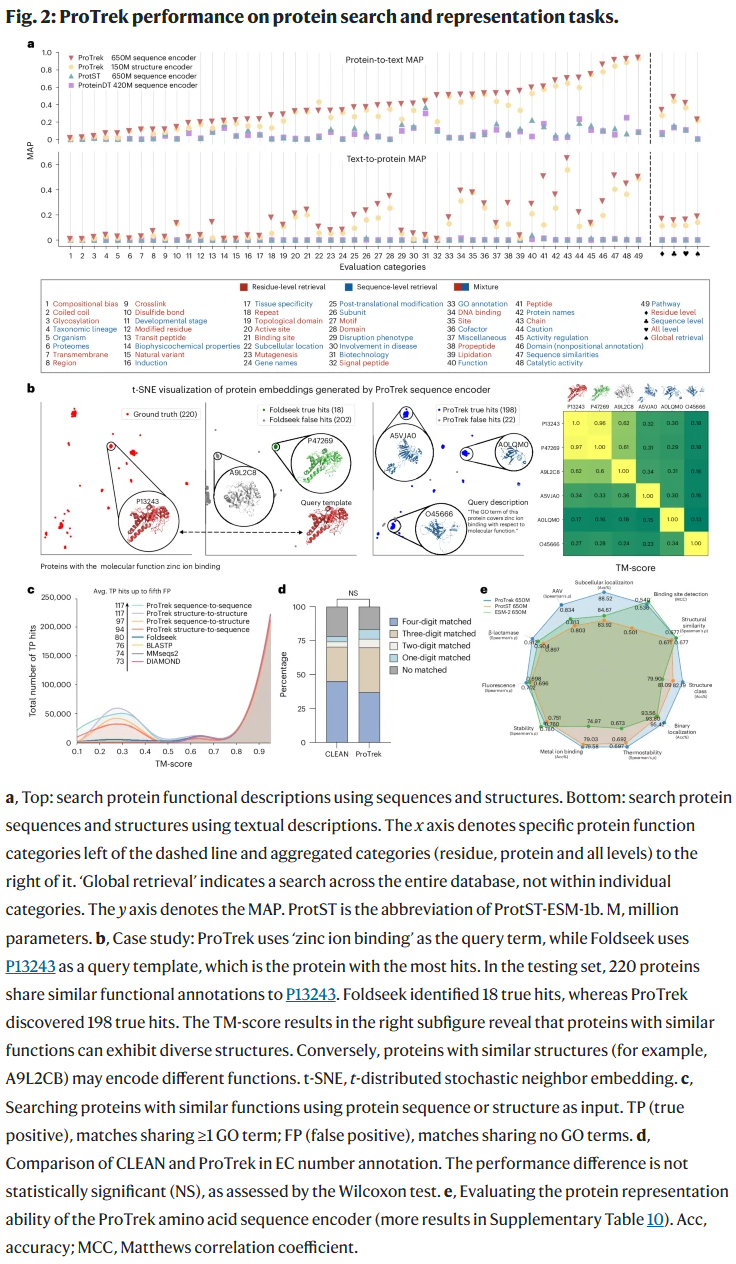
ProTrek 支持九类检索任务(序列↔结构、序列↔功能、结构↔功能,以及单模态内部检索)。在 SWISS-PROT 测试集上,ProTrek 显著优于 ProteinDT 和 ProtST,MAP 指标提升超过 30-60 倍,尤其在全局检索任务中优势突出。
文本-蛋白互译
在基于文献的验证实验中,ProTrek 能准确将文本描述映射到目标蛋白,或将蛋白序列检索到正确的功能描述。即使遇到训练集中未包含的新酶或 CRISPR 蛋白,ProTrek 依然能正确识别,展示出较强的泛化能力。
蛋白功能收敛进化识别
通过比对酶活性和 CRISPR 蛋白的功能文本,ProTrek 能识别出功能类似而序列或结构差异显著的蛋白,揭示了收敛进化现象。这是传统基于同源性的方法难以实现的。
与比对工具的对比
在序列-序列、结构-结构等任务中,ProTrek 超越了 BLASTp、MMseqs2 和 Foldseek,尤其在低相似度区域(“暮光区”)仍能准确找到功能相似的蛋白。
实验验证
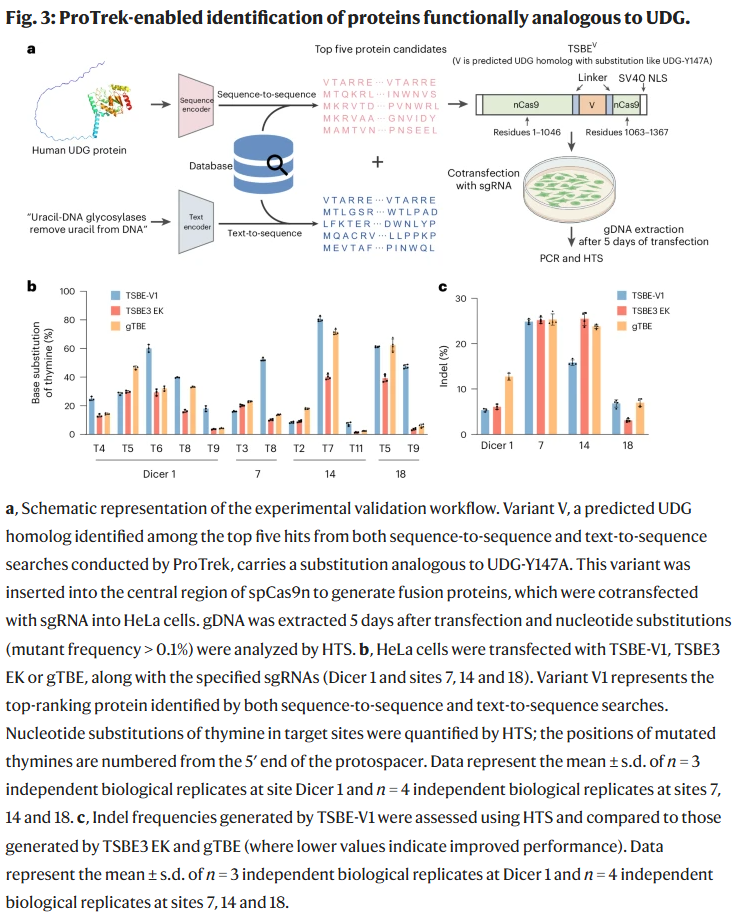
研究人员利用 ProTrek 搜索到一批与人类 UDG 蛋白功能类似的候选蛋白,并通过突变设计出新的胸腺嘧啶切除酶,提升了碱基编辑效率。这些蛋白在细胞实验中表现出比现有编辑器更高的效率和更低的脱靶率。
推理速度与可扩展性
基于最大内积搜索算法,ProTrek 能在数秒内完成亿级数据库检索,比现有工具快百倍以上。目前 ProTrek 已整合 50 亿蛋白,未来目标是覆盖 100 亿条。
讨论
ProTrek 在统一蛋白序列、结构和功能模态方面展现出突破性能力,不仅能完成高效准确的搜索,还能支持收敛进化分析、功能注释和新蛋白发现。其优势在于:
跨模态表示学习,打破单模态限制。
强大的文本理解能力,支持自然语言检索。
高效的搜索速度,适合超大规模蛋白数据库。
尽管 ProTrek 在新设计蛋白或细微序列变异上仍存在不足,但研究人员认为它已成为生成生物学假设、发现新蛋白和探索蛋白功能模式的有力工具。
整理 | DrugOne团队
参考资料
Su, J., He, Y., You, S. et al. A trimodal protein language model enables advanced protein searches. Nat Biotechnol (2025).
https://doi.org/10.1038/s41587-025-02836-0

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢