
DRUGONE
自由能计算是量化药物与靶标结合亲和力的关键工具。结合自由能反映了配体与受体结合时体系能量的变化,是药物筛选与优化中的核心指标。虽然自由能理论框架早在几十年前已建立,但其在药物-靶标体系中的高效应用仍是计算化学的重要挑战。
研究人员回顾了两大类增强采样方法的进展与挑战:炼金转化法(alchemical transformations)与基于路径的方法(path-based methods)。炼金方法能有效计算能量差,是制药工业中最常用的相对结合自由能计算手段,但在绝对自由能预测及结合机制解析上仍有限。而基于路径的方法不仅能获得绝对自由能,还能揭示结合途径与动力学特征。本文比较了这两类方法的原理与适用场景,重点介绍了结合机器学习发展的新型路径采样策略,以及研究团队基于MetaDynamics与非平衡模拟的半自动化自由能计算流程,并讨论平衡与非平衡计算方案的优势与局限。

药物候选物的筛选往往依赖多个参数,其中结合亲和力是最重要的指标之一。实验上可通过表面等离子共振、等温滴定量热法、放射或酶学实验测定结合常数。然而,随着计算能力(尤其是GPU计算)的快速提升,计算结合亲和力成为计算药物设计中降低成本与加速研发的重要途径。
结合亲和力与结合自由能直接相关,后者可通过分子动力学(MD)模拟精确估计。自由能计算的基础源自统计力学理论,发展至今主要形成两类路径:
炼金转化法(Alchemical transformations):通过非物理参数平滑连接初态与终态;
基于路径法(Path-based methods):沿实际反应坐标或集体变量计算势能面与平均力势。
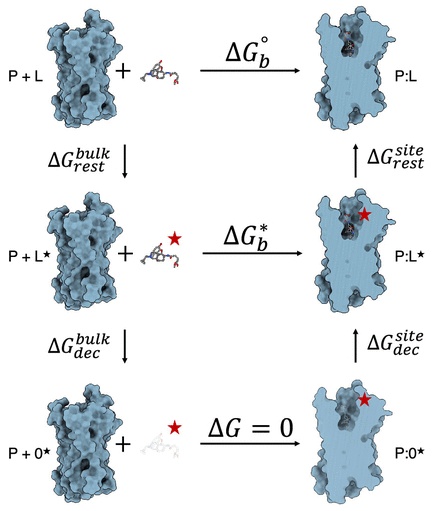
图1. 炼金双解耦热力学循环示意图
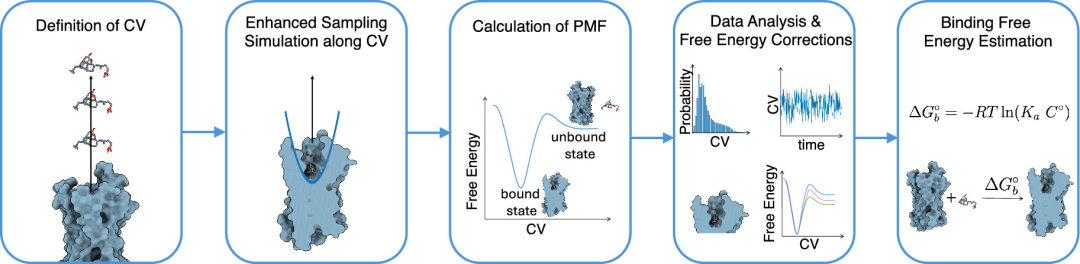
图2. 基于路径的结合自由能计算工作流程
集体变量与耦合参数
在炼金转化中,引入耦合参数λ以描述体系从初态到终态的平滑变化;而在路径法中,体系演化通过集体变量(CV)刻画,这些变量可代表距离、角度、均方差或更复杂的体系自由度。
设计高效CV对于捕捉体系动力学至关重要。对于复杂体系(如蛋白-配体结合),简单几何CV常不足以描述真实动力学,因此研究人员提出了路径集体变量(Path Collective Variables, PCVs),用于在高维空间中追踪体系沿预定义路径的演化。
基于PCV的自由能计算能准确描述配体结合与大尺度构象变化,是连接统计力学理论与药物发现实践的重要桥梁。
炼金转化法
炼金方法包括自由能微扰(FEP)和热力学积分(TI),两者均利用非物理路径连接体系初态与终态。
在药物设计中,炼金方法常用于估算相似化合物的相对结合自由能,是制药企业优化先导分子的标准手段。
炼金计算通过混合哈密顿函数在不同λ值下分层采样,计算能量差。最早应用于小分子体系,后扩展至复杂配体如抗癌药伊马替尼(Gleevec)的结合研究。
研究人员指出,炼金法在精度上已能达到亚千卡水平,但其缺陷在于:
仅能提供能量差,缺乏结合机制信息;
对大幅结构变化体系收敛困难;
计算复杂度高,参数依赖显著。
为改进这一问题,研究人员提出结合双向采样的统计方法(如BAR方法),通过正反向转化提升收敛性。炼金转化法已在溶剂化自由能、相对稳定性及晶态与非晶态药物可溶性分析中取得广泛应用。
基于路径的方法
伞形采样(Umbrella Sampling, US)
为解决标准MD难以跨越高能垒的局限,US在1977年被提出,通过在不同集体变量区间施加谐波偏置势增强采样。 US能够有效重建平均力势(PMF),并揭示离子对、蛋白构象变化或配体结合的能量曲面。研究人员利用US结合PCV探索过渡态类似物与酶的结合路径,并通过机器学习算法(高斯核正则回归)平滑重建自由能面,实现高精度拟合。然而,US方法需精确选择偏置强度与窗口分布,操作复杂,限制了其在药物工业中的广泛应用。
元动力学(MetaDynamics, MetaD)
MetaD通过不断添加高斯势项阻止体系返回已采样区域,从而推动体系跨越能垒并探索自由能地形。
改进后的调温元动力学(Well-Tempered MetaD)可平滑收敛并减少偏置误差。研究人员广泛使用MetaD分析配体结合与解离机制、精修对接构象,并在药物设计中结合机器学习开发了半自动化自由能计算流程。该流程显著减少了参数数量与计算时间,在预测蛋白激酶配体结合能方面表现出优异的实验一致性,代表了MetaD方法在药物自由能研究中的最新进展。
受力分子动力学(Steered Molecular Dynamics)
SMD通过对配体施加外力模拟结合或解离过程,是典型的非平衡增强采样方法。研究人员通过分析配体拔出轨迹的受力曲线与能量耗散,揭示关键结合与断裂事件,可区分活性与非活性抑制剂。
最新工作中,SMD结合PCV并基于Crooks涨落定理进行双向自由能估计,不仅提高了准确度,也能揭示详细的分子解离路径。SMD的优势在于易于并行化,可同时运行多个轨迹以加速收敛,并可与机器学习结合识别关键结合特征。
挑战与展望
自由能计算在药物发现中已取得重要突破,但仍存在三大挑战:
采样困难:结合事件属于稀有过程,常需耗时巨大的MD采样;
力场精度有限:传统力场难以准确描述复杂的相互作用;
收敛性问题:非平衡模拟与元动力学中常存在收敛偏差。
研究人员指出,未来改进方向包括:
发展机器学习驱动的集体变量以提高采样效率;
引入神经网络力场(如OBIWAN模型)改善能量面精度;
建立标准化的收敛评估准则,确保结果稳定与可重复。
此外,新方法如炼金转移法(Alchemical Transfer Method, ATM)与哈密顿复本交换(H-REMD)结合了炼金与路径两类思路,兼顾计算效率与物理可解释性。最终目标是将自由能计算系统性地整合进药物研发流程,从传统的静态分子对接过渡到动态对接与自由能引导设计,实现更准确的结合能预测与先导优化。
整理 | DrugOne团队
参考资料
On Free Energy Calculations in Drug Discovery. Alessia Ghidini, Eleonora Serra, and Andrea Cavalli. Accounts of Chemical Research.
DOI: 10.1021/acs.accounts.5c00465

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢