
DRUGAI
近年来,AI在药物研发中的应用日益广泛,尤其是在分子表征与虚拟筛选领域。由厦门大学、北京协和医学院与北京大学联合研究团队开发的Geometry-based BERT(GEO-BERT)模型,在多个分子性质预测任务中取得了领先性能,并在药物筛选实践中成功发现了两种高活性的新型DYRK1A抑制剂。该成果展示了AI模型在早期药物发现中的应用潜力。

研究背景
传统药物发现依赖高通量筛选技术,但该方法成本高、效率低,严重制约新药研发进程。随着计算技术的发展,虚拟筛选成为加速药物发现的重要手段,其中分子性质预测模型是核心工具之一。早期方法多依赖人工设计的分子指纹或描述符,存在特征工程复杂、泛化能力弱等问题。近年来,深度学习模型如RNN、CNN和GNN被广泛应用于分子表征,但受限于标注数据不足,模型性能仍有提升空间。BERT模型因其自监督预训练机制和双向注意力结构,在分子性质预测中展现出良好性能。然而,现有的BERT类模型多数是基于分子的二维结构开发的,忽略了三维构象对于分子发挥活性的重要性。
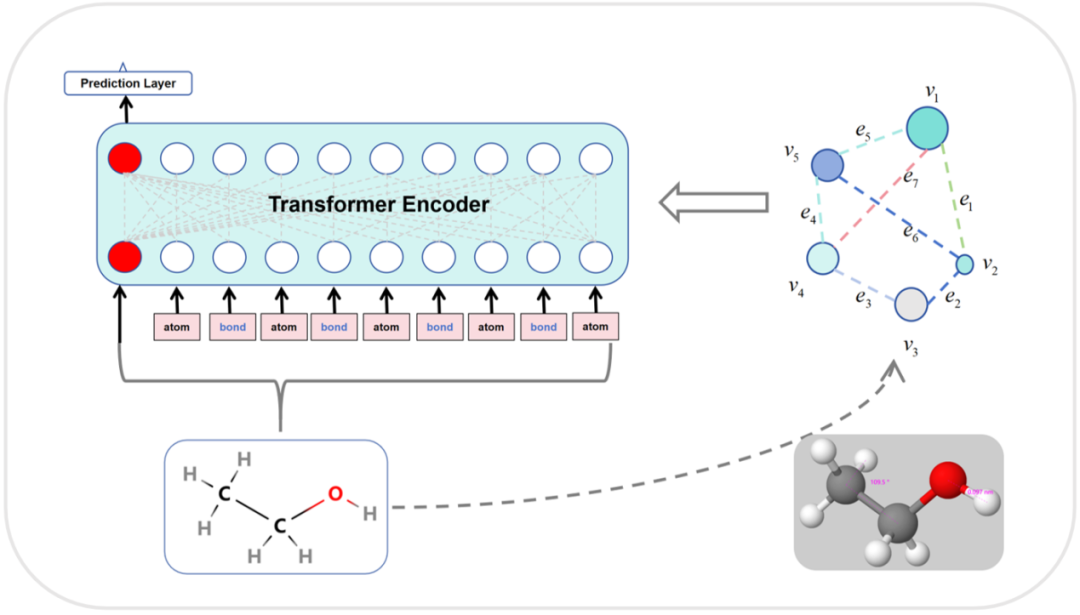
图1 GEO-BERT示意图
研究内容
为突破现有模型的局限,研究团队提出了GEO-BERT模型,将分子的三维几何信息(原子间距离、键角等)引入Transformer架构中。该模型以原子和化学键作为独立分词(Token),构建了三种连接关系:原子-原子、键-键、原子-键,并在注意力机制中引入三种空间矩阵:原子距离矩阵、键角矩阵和原子-键邻接矩阵。通过这种设计,模型能够在每一层注意力计算中同时关注分子的语义结构与空间构象,从而提升分子表征的准确性与泛化性能。
在预训练(Pre-training)阶段,模型使用来自ChEMBL数据库的145万个类药性分子数据,并通过RDKit生成最低能量构象以提取三维信息。在微调阶段,模型在8个公开数据集(如Tox21、ClinTox、HIV等)上进行迁移学习。在真实药物筛选实践任务中,模型针对双特异性酪氨酸磷酸化调节激酶1A(DYRK1A)靶点进行迁移学习,并应用于DYRK1A抑制剂的发现。
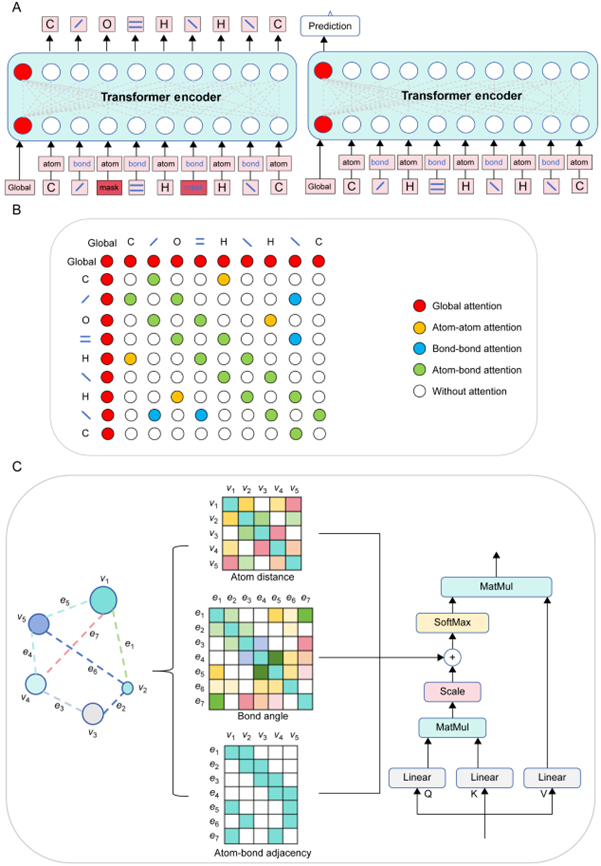
图2 GEO-BERT架构
🧪 研究结果
在基于8个公开数据集的基准测试中,GEO-BERT在多个数据集上均优于现有主流模型(如FG-BERT、MoleBERT),在AUC、F1-score、MCC等指标上表现突出。 研究引入了SRD(Sum of Ranking Differences)方法对模型性能进行综合评估,结果显示GEO-BERT在多个任务中排名稳定,表现优异,具备良好的一致性。
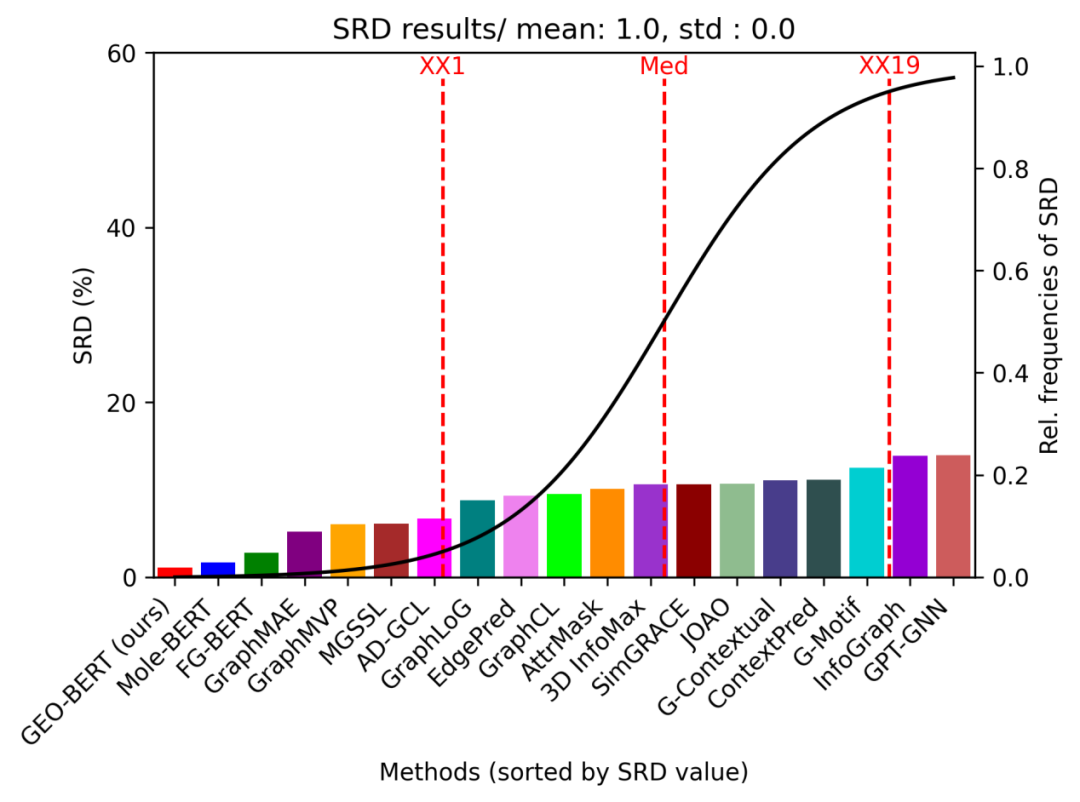
图3 GEO-BERT的SRD排名
研究团队以药物筛选实践任务(DYRK1A)为例对GEO-BERT模型的多种性能进行理论分析。
(1)模型的可解释性。如图3所示,。研究团队应用t-SNE技术分别展示了不同原子类型、化学键类型以及原子簇的嵌入向量分布情况,揭示了模型在学习分子结构时的区分能力。此外,研究团队还展示了模型在代表性分子上的注意力分布,表明GEO-BERT能够有效识别分子中关键原子或原子簇,为后续药效机制研究提供了依据。如图4所示,研究团队选取了数据集中的活性分子与非活性分子并进行对比分析,结果显示模型在能够准确定位活性分子中具有代表性的官能团或关键结构片段,也可呈现出非活性分子的不同于活性分子注意力分布。这一发现不仅验证了模型的判别能力,也为药物设计中的结构优化提供可视化参考。
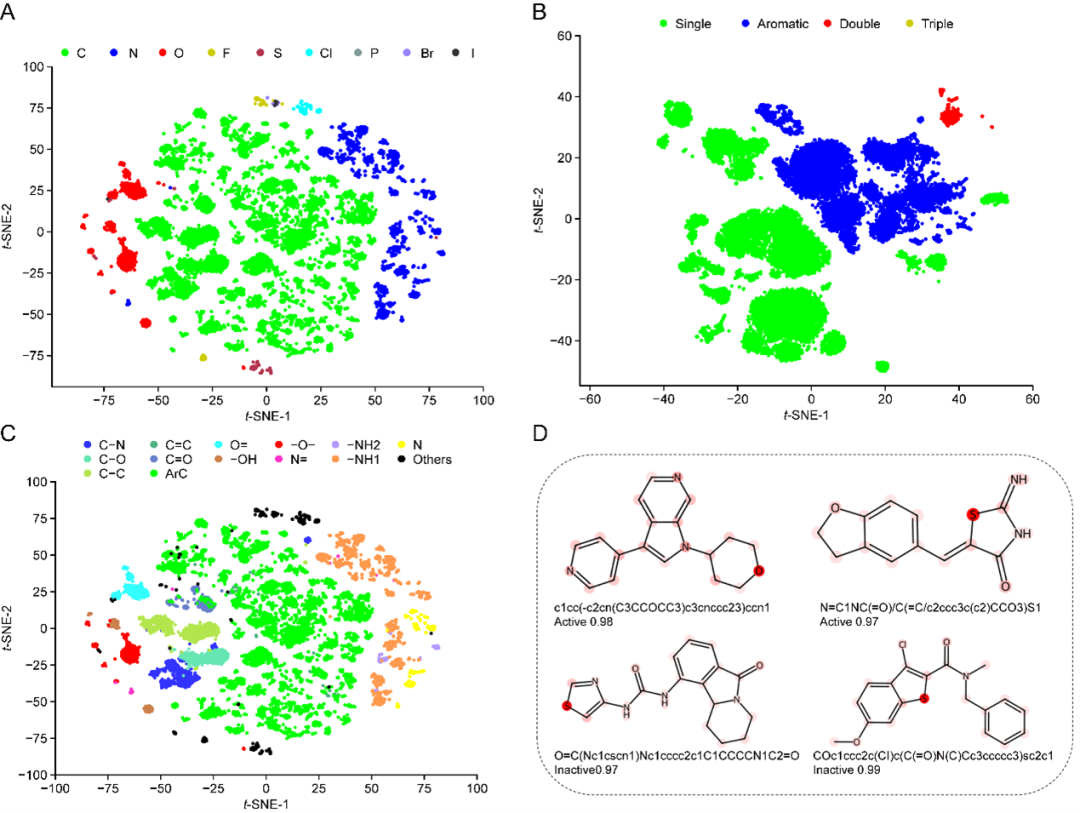
图4 GEO-BERT的t-SNE分析
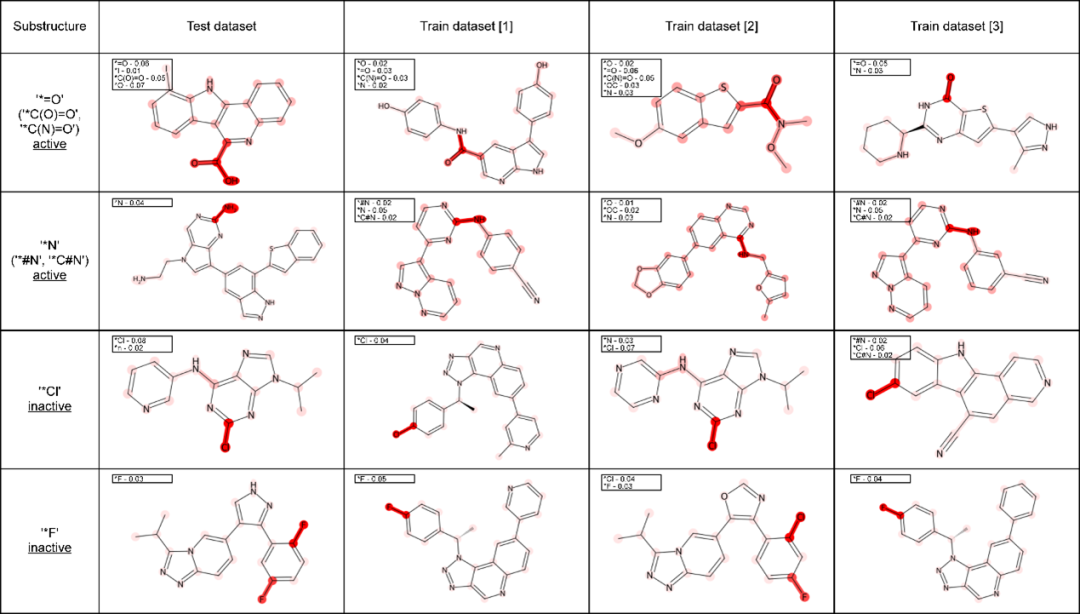
图5 GEO-BERT的子结构可解释性
(2)模型可靠性评估。研究团队对GEO-BERT的不确定性进行了系统分析。研究采用熵、蒙特卡罗丢弃法以及集成方法三种策略,量化模型在不同预测置信度下的表现。结果表明模型在低不确定性区间内预测更为准确,而高不确定性区间则提示潜在的误判风险。该分析为后续药物筛选中的风险控制与决策支持提供了重要依据。
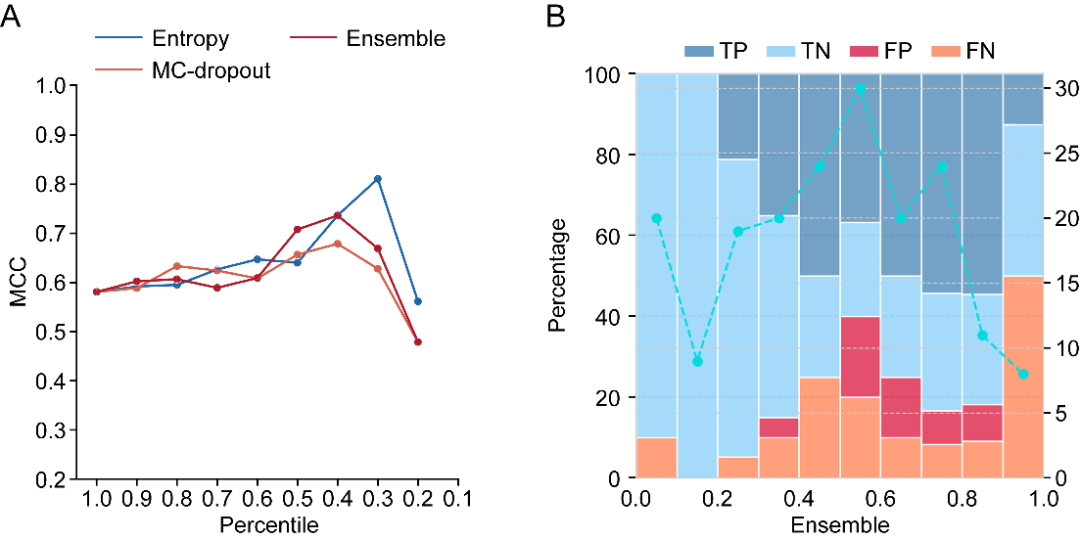
图 6 GEO-BERT的不确定性分析
为验证模型的实际应用价值,研究团队GEO-BERT在DYRK1A数据集迁移学习获得的模型应用于高通量药物筛选。模型从SPECS化合物库(20万)中快速预测到500个潜在活性分子。通过分子对接辅助,进一步选出20个最具潜力的化合物。经过实验验证,最终发现2个亚微摩尔级别的新型DYRK1A抑制剂,证明了GEO-BERT在真实药物发现实践中的实用性。
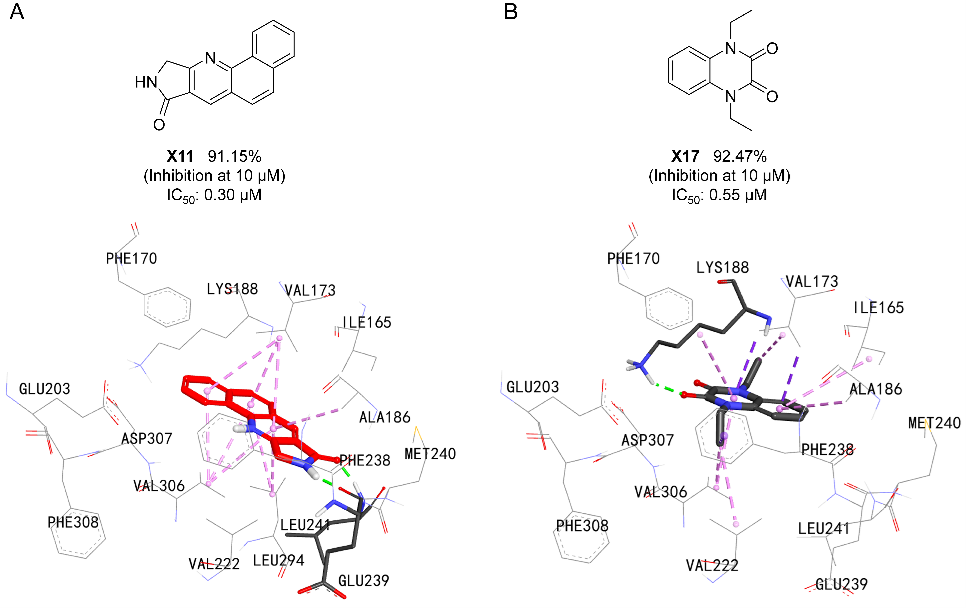
图 7 GEO-BERT发现的两种DYRK1A抑制剂
研究团队进一步利用t-SNE方法对新发现的DYRK1A抑制剂进行了可视化展示。图8中不同颜色的点代表不同类型的预测结果,点的大小则表示每个样本的不确定性值(由集成方法计算)。该图不仅揭示了模型预测高活性分子X11与X17的可靠性,也表明两个分子的结构区别于已知分子因而具有结构新颖性。
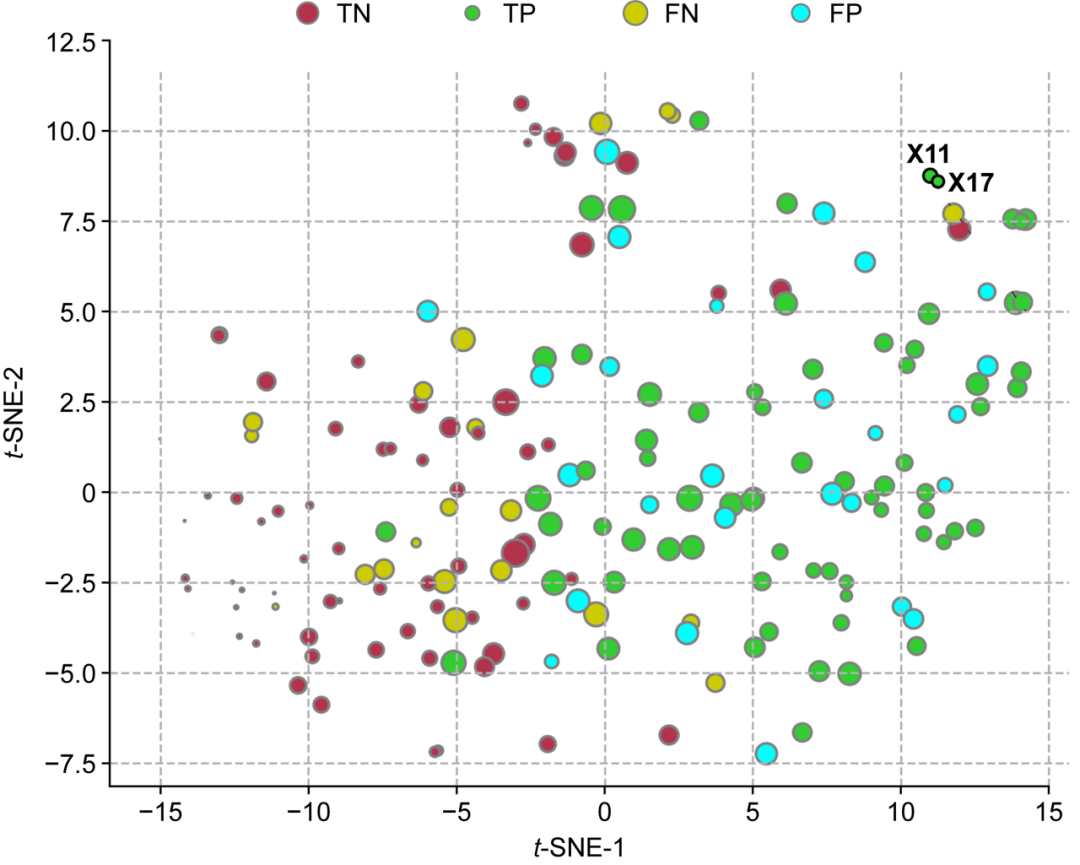
图8 2个新型抑制剂X11与X17与已知抑制剂分子的空间分布(t-SNE方法)
总结与展望
GEO-BERT模型通过引入分子的三维几何信息(包括原子间距离、键角和原子-键邻接关系),在Transformer架构中构建了多维注意力机制,从而显著提升了分子表征的精度与泛化能力。该模型在多个公开数据集上表现优异,全面优于现有主流BERT类分子表征模型,并在真实药物筛选任务中成功发现了两种高活性的新型DYRK1A抑制剂,验证了其在实际药物研发中的应用价值。
此外,研究团队还从模型可解释性与可靠性角度进行了深入分析,包括原子与键嵌入向量的t-SNE可视化、注意力分布图、子结构识别能力、不确定性评估等,全面展示了GEO-BERT在分子识别、活性预测和风险控制方面的良好性能。这些分析不仅增强了模型的透明度,也为后续药物设计与筛选提供了结构层面的参考依据。未来,GEO-BERT有望在高通量药物筛选、全新分子设计成药性预测等新药研发领域发挥重要作用,推动人工智能在制药工业中的深度融合与落地应用。
参考资料
Xiang Zhang, Chenliang Qian, Bochao Yang,Hongwei Jin, Song Wu,Jie Xia*, Fan Yang* and Liangren Zhang. Geometry-based BERT: An experimentally validated deep learning model for molecular property prediction in drug discovery. Journal of Pharmaceutical Analysis. 2025: 101465.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢