
DRUGONE
冷冻电镜(cryo-EM)与X射线晶体学为解析生物大分子的原子级结构提供了关键数据。现有的精修过程依赖基于库的立体化学约束,但这些约束不仅局限于已知化学实体,还缺乏对非共价相互作用的真实刻画。量子力学(QM)计算能够解决这些问题,但计算代价极高。研究人员提出了一种新型AI辅助量子精修方法——AQuaRef(AI-enabled Quantum Refinement),其基于机器学习原子间势能模型AIMNet2,能以远低于传统量子计算的代价实现量子级精度。AQuaRef在41个cryo-EM与30个X射线结构的精修中表现出显著优越的几何质量,同时保持甚至提升了实验数据拟合度。尤其在分析帕金森病相关蛋白DJ-1及其细菌同源物YajL的短氢键结构时,AQuaRef成功辅助确定质子位置,展示了其在复杂化学环境中的精确性与可靠性。

尽管AlphaFold、RoseTTAFold等预测模型为结构生物学提供了革命性工具,但实验结构解析仍是理解分子功能与药物设计的基石。
在结构精修阶段,研究人员旨在在尽可能符合实验数据的同时,确保分子几何合理。当前的精修软件(如Phenix、CCP4)依赖标准化学库约束原子间几何参数(键长、键角、手性等),以防止结构畸变。然而这些约束存在多重局限:
仅适用于已知化学实体;
无法准确反映氢键、π堆叠等非共价相互作用;
难以处理异常键长或复杂共价偏离;
新配体或交联结构需人工手动注释。
量子精修(Quantum Refinement)通过将实验数据拟合项与体系的量子能量项同时优化,能从物理本源刻画几何约束。然而,全分子量子精修在历史上因计算成本过高而不可行。AQuaRef借助机器学习势能模型(MLIP)实现了首次在蛋白质整体尺度上的量子精修。
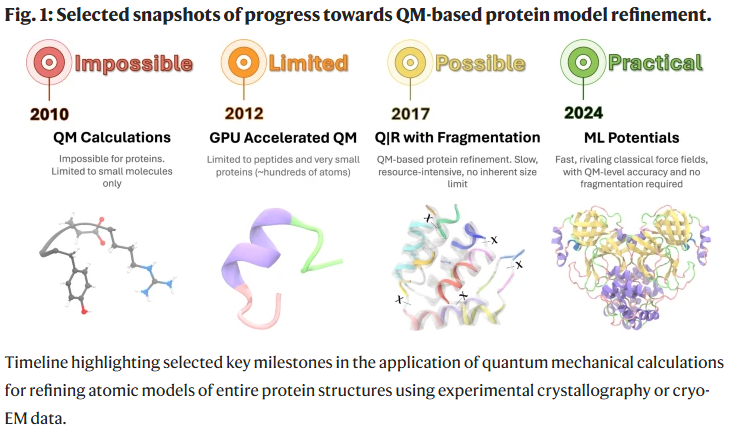
图1|蛋白质量子精修的发展历程
方法
AIMNet2模型与量子势能设计
AQuaRef采用AIMNet2神经网络势能,其具备旋转不变特征、消息传递机制与总电荷处理能力。研究人员基于多肽体系构建了训练集,涵盖20种标准氨基酸、不同质子化状态及多肽二硫键/硒键组合,并通过隐式溶剂修正获得量子级能量标注。最终模型在百万级样本上训练,达到近似B97M-D4/def2-QZVP量子精度。
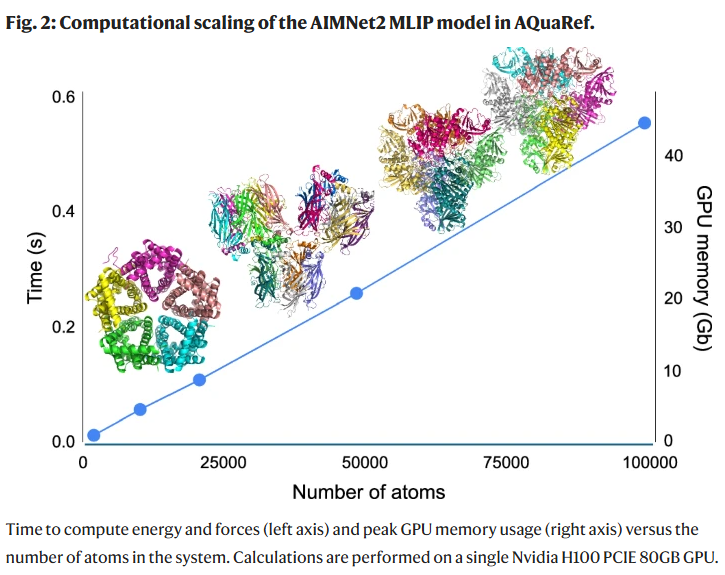
图2|AIMNet2计算扩展性
AQuaRef量子精修流程
AQuaRef通过Q|R软件模块实现,流程包括:
检查并补全原子(含氢);
对晶体结构扩展晶胞并修正对称性;
进行几何正则化以消除空间冲突;
使用AIMNet2量子势能计算结构能量与梯度;
迭代最小化残差以获得最优原子模型。
平均精修时间仅为标准方法的两倍(多数模型<20分钟),可在普通GPU笔记本上完成。
结果
低分辨率模型的系统改进
对61个蛋白质结构(41个cryo-EM、20个X射线)进行测试后发现:
AQuaRef显著提升MolProbity评分、Ramachandran Z分数与CaBLAM几何一致性;
与实验数据拟合度(CCmask、Rfree)保持或略有提升;
在X射线模型中,Rwork–Rfree差距减小,表明过拟合降低;
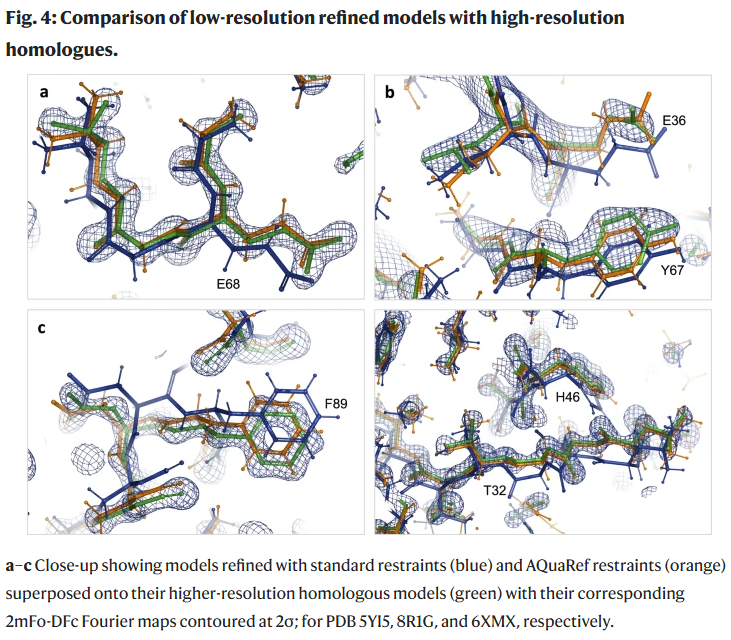
与高分辨率同源结构对比,AQuaRef精修后模型更接近真实几何。
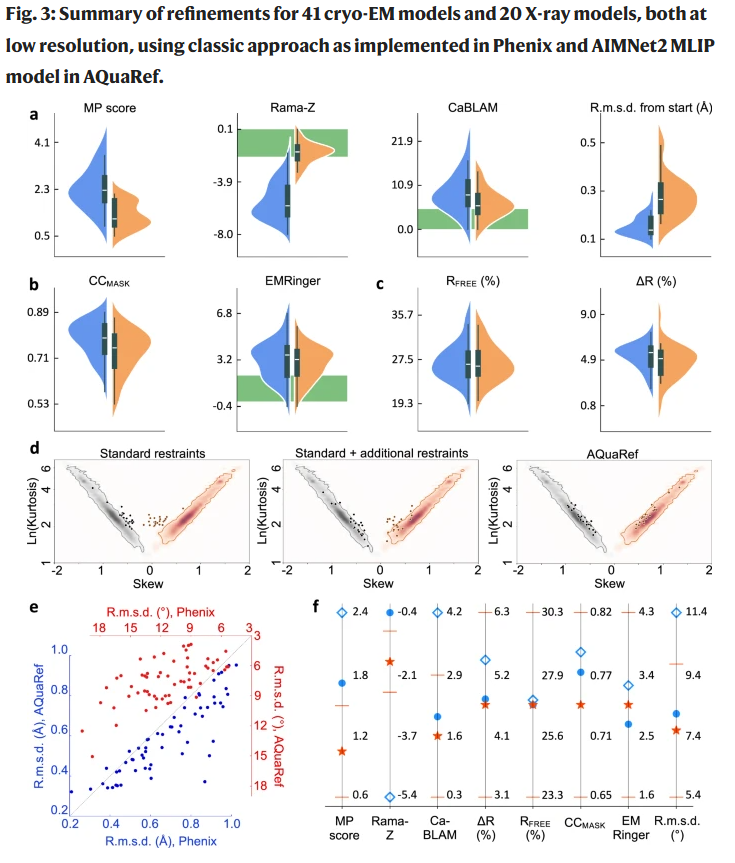
图3|AQuaRef与标准精修几何性能对比
与其他精修软件的比较
研究人员将AQuaRef与REFMAC5、AMBER、Rosetta及Servalcat进行系统比较:
X射线模型:AQuaRef的Rfree略优,且Rfree–Rwork差最小;
cryo-EM模型:AQuaRef与Rosetta几何质量相当,显著优于Servalcat;
整体几何:AQuaRef和Rosetta在MolProbity与CaBLAM指标上最佳。
Rosetta的非梯度采样优化在局部结构上略胜一筹,但计算耗时约AQuaRef的10倍。
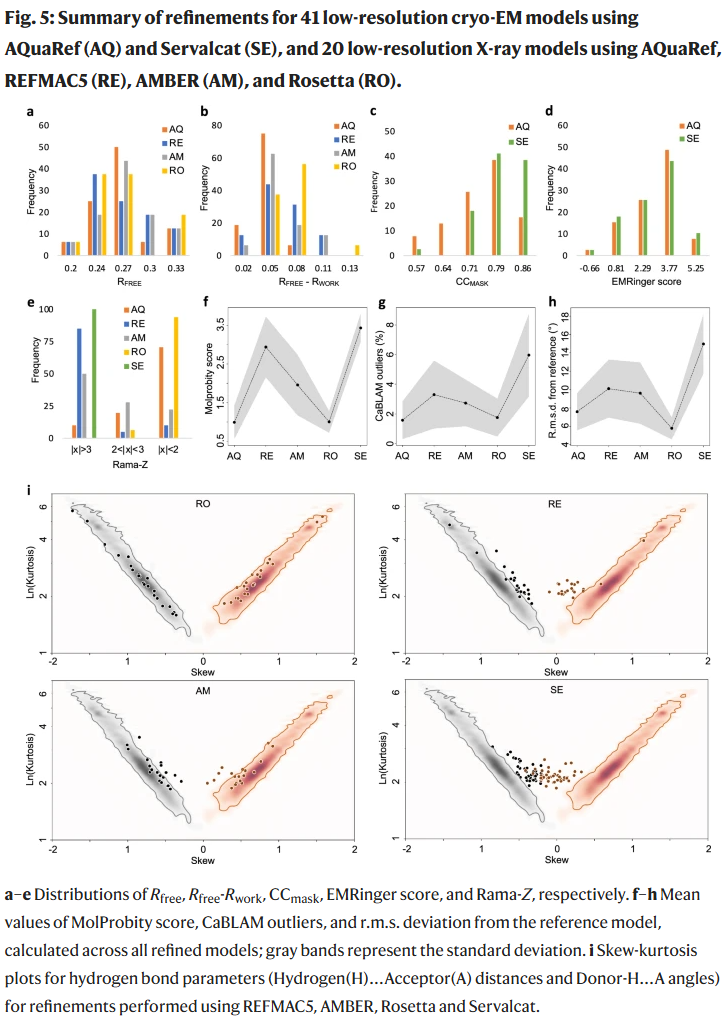
图5|AQuaRef与其他主流精修算法性能比较
案例分析:DJ-1与YajL短氢键结构
质子定位是高分辨率结构精修中的关键挑战。研究人员以DJ-1与其细菌同源物YajL为例,验证AQuaRef在亚埃分辨率下的可靠性。
DJ-1:AQuaRef精修自动识别E15–D24之间的共享质子位置,与实验结果一致;
YajL:AQuaRef揭示E14–D23之间的低势垒氢键(LBHB),质子分布于Oε2与Oδ2之间。
AIMNet2能量剖面与差值密度图均支持该解释。
在2 Å截断数据下,AQuaRef仍准确恢复质子位置,而传统约束方法产生偏差。
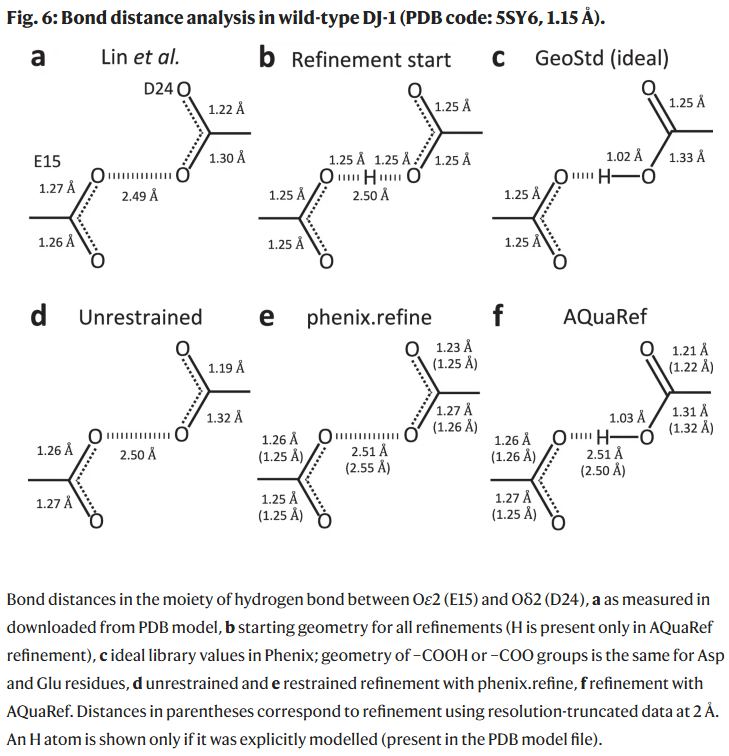
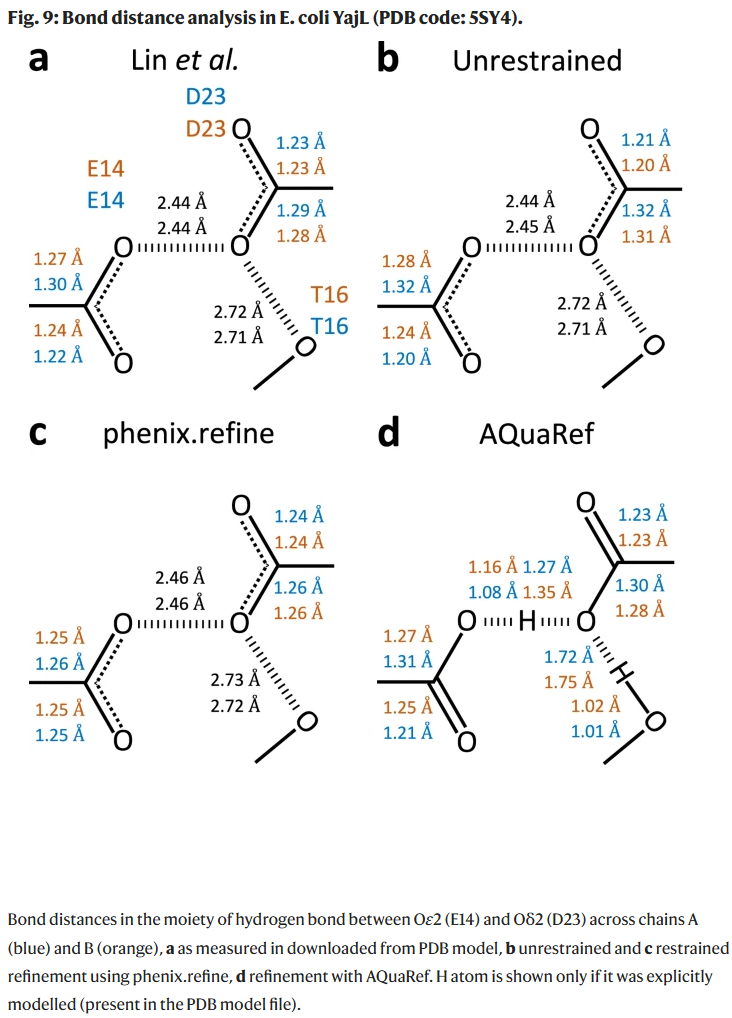
讨论
AQuaRef实现了首个基于机器学习势能的全蛋白量子精修:
显著提升几何合理性与验证指标;
在低分辨率下减少数据过拟合;
能准确定位质子与氢键网络;
保持较低计算代价与高适用性。
该方法已集成至Phenix的Q|R模块,向结构生物学界开放使用。
未来工作包括:
扩展至含配体与核酸体系;
引入对静态无序与多构象的处理;
优化模型泛化性与量子–机器学习混合精度。
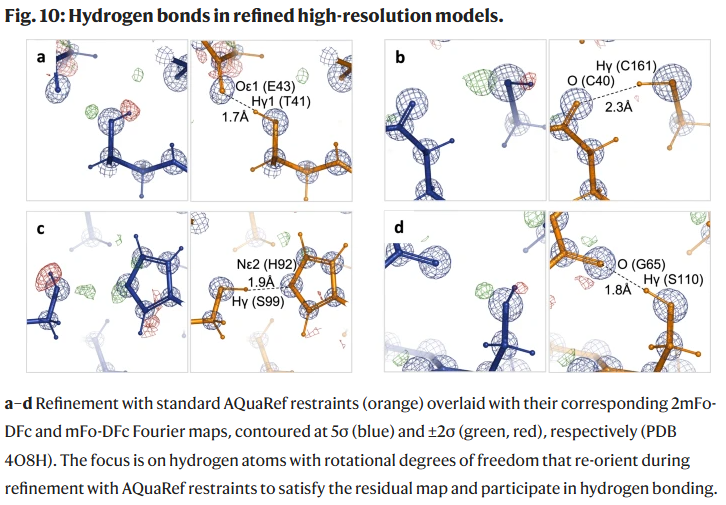
图10|高分辨率结构中氢键的重定位可视化
结论
AQuaRef标志着结构生物学进入“机器学习量子精修”时代。它以量子精度、机器学习速度和高可解释性实现了全蛋白原子模型的优化。该框架不仅提升了结构质量,更在理解非共价相互作用、质子转移和反应性中心方面,为未来的分子模拟与药物设计提供了强大工具。
整理 | DrugOne团队
参考资料
Zubatyuk, R., Biczysko, M., Ranasinghe, K. et al. AQuaRef: machine learning accelerated quantum refinement of protein structures. Nat Commun 16, 9224 (2025).
https://doi.org/10.1038/s41467-025-64313-1

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢