
本文介绍一篇来自浙江大学侯廷军、谢昌谕团队联合发表的研究成果“Biology-driven insights into the power of single-cell foundation models”。该研究对当前六种主流的单细胞基础模型进行了系统性的基准评估,涵盖基因层面与细胞层面的多个任务,并引入基于细胞本体论的新型评估指标,揭示了scFMs在生物学任务中的真实表现与适用范围,为模型选择与应用提供了实用指南。

研究背景
近年来,单细胞RNA测序(scRNA-seq)技术的发展极大地拓宽了我们对生物学过程和疾病机制的理解。随着高通量测序技术的进步,单细胞转录组数据量呈指数级增长,但其固有的高维、稀疏和低信噪比等特点,给传统机器学习模型的应用带来了挑战。受自然语言处理(NLP)领域基座模型成功的启发,基于Transformer架构的单细胞基础模型(scFMs)应运而生,旨在通过大规模预训练来学习通用的生物学知识,有望在多种下游任务中实现零样本或少样本的高效迁移。然而,scFMs是否能够超越标准方法提取独特的生物学见解尚不明确,其在细胞分类等特定任务中的优势也受到一些工作的质疑。因此,迫切需要一项全面的基准研究来指导scFMs的构建和实际应用。
基准框架概览
为了解决上述问题,研究人员构建了一个生物学应用驱动的基准框架,对当前主流的六个scFMs(Geneformer、scGPT、UCE、scFoundation、LangCell和scCello)的零样本嵌入能力(Zero-shot embeddings)进行了深度评估,涵盖以下3个层面的7类任务:
基因层面任务:组织特异性基因预测、基因功能预测;
细胞层面任务:批次整合(细胞聚类)、细胞类型注释、癌细胞识别、药物敏感性预测;
可解释性分析:基于注意力分数的基因调控关系分析。
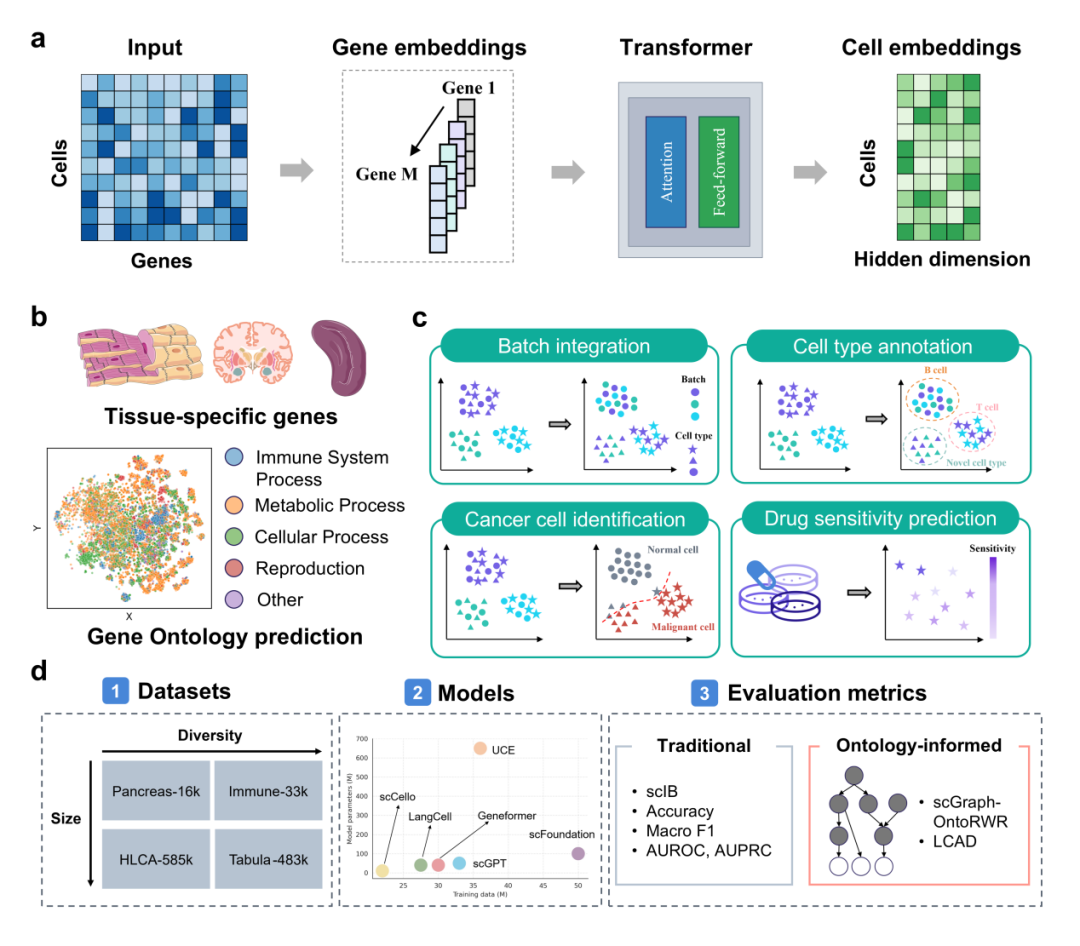
图1. scFM-Bench基准评估框架概览图
结果与讨论
批次整合: scFMs更善于保留生物学结构
本研究在5个不同规模和复杂度的数据集上进行了批次整合测试。为了避免数据泄漏的影响,研究人员选取了2025 年 4 月发布的 AIDA v2 数据集中新增的约 201K个健康供体免疫细胞,由于该数据首次公开时间晚于scFMs的发布时间,可以确保该数据不存在于任何scFM的预训练数据中。
传统评估指标(scIB)显示,scFMs并未超越简单的基线方法,如高可变基因(HVG)、scVI(图 2a)。然而,有研究表明scIB指标过高的模型可能破坏了数据本身的生物学结构,并据此提出了scGraph指标,用于衡量表征空间中细胞类型间远近关系与原始基因表达数据的一致性。scGraph-PCA指标根据原始基因表达的PCA降维结果来构建参考图,这意味着用于评估表征空间质量的参考图会随着数据集的变化而变化。本研究进一步提出了 scGraph-OntoRWR指标,旨在提供一种稳健的基准分析方法。该指标通过以下步骤实现:(1)将细胞本体论(Cell Ontology)建模为有向无环图(DAG),图中节点间的连线表示细胞的从属关系;(2)在图上执行带重启的随机游走算法(Random Walk with Restart, RWR),计算出平衡分布来量化细胞类别的内在关联度,从而生成一个不依赖于特定数据集的参考图,用于稳健且一致的细胞间距离计算。
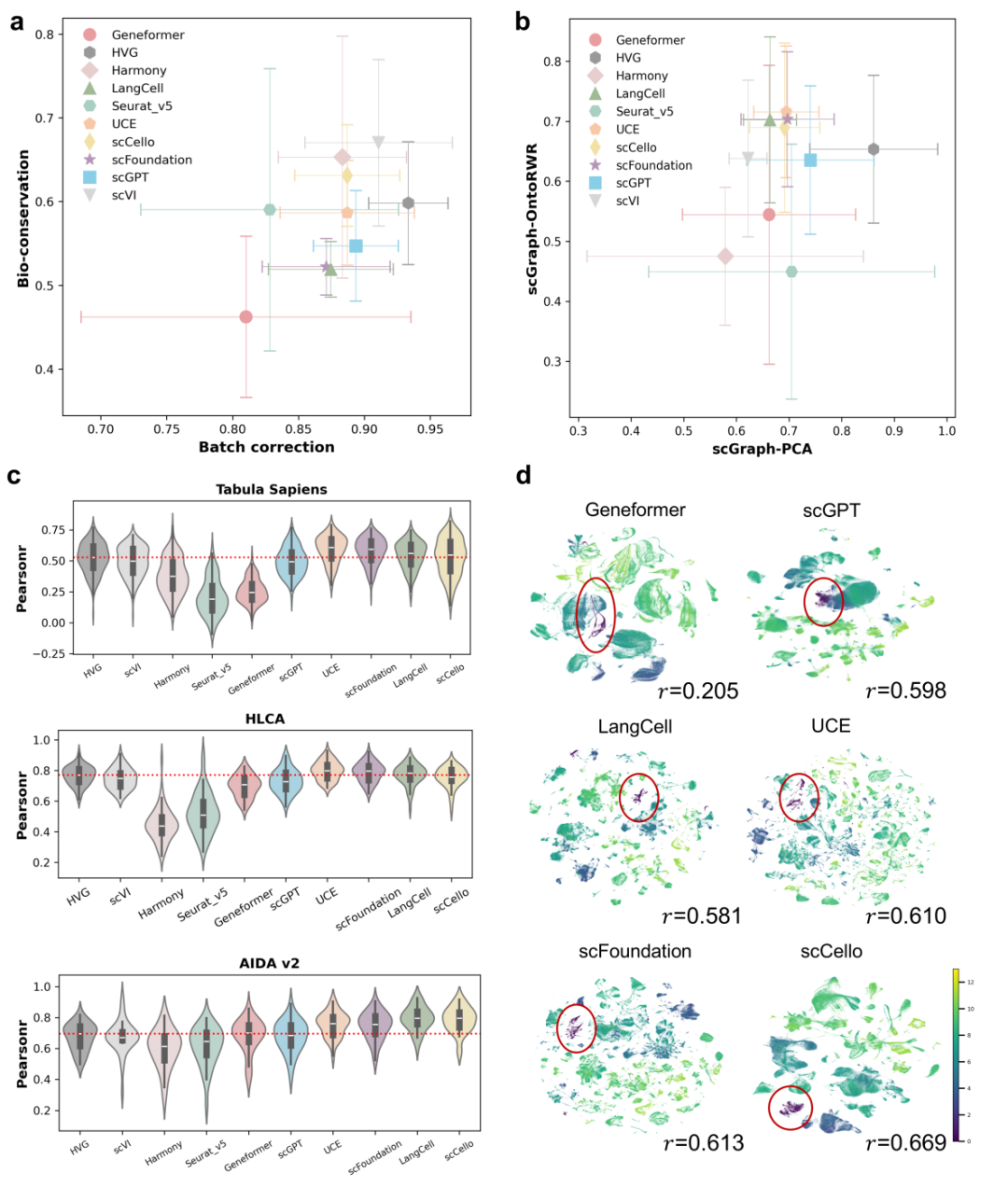
图2. 批次效应整合实验结果
实验结果表明,scFMs 在 scGraph-OntoRWR 指标下超越了基线方法(图 2b)。平均而言,UCE 模型表现最优,其次是 scFoundation 和 LangCell。以每种细胞类型为锚点,研究人员计算并可视化了细胞类型特异性 scGraph-OntoRWR 分数的分布(图 2c),突出了 LangCell 和 scCello 在无偏 AIDA v2 数据集上的优越性。scGraph-OntoRWR指标通过提供生物学感知视角对传统指标进行了有力补充,并证明了scFMs能够更好地保留细胞类型之间的生物学关系,尤其是在跨组织、跨样本的复杂数据中。
细胞分类: scFMs具有跨数据集和分布外可迁移性
本研究评估了 scFMs 在细胞类型注释中的性能,包括数据集内验证、跨数据集验证和新细胞类型的识别三大场景。为了更全面地评估模型性能,本研究从细胞类别间层次结构出发,设计了两个本体论相关指标:Accuracy (non-leaf) 和最近公共祖先距离(lowest common ancestor distance, LCAD)。
在训练细胞分类模型时,研究人员通常会将带有粗粒度标签的数据剔除,以确保最终的细胞类别是互相独立的。比如,如果“T-cell”和"CD4+ T-cell"同时存在于数据集中,“T-cell”会被剔除。本研究将这部分被排除的粗粒度数据重新纳入,用于额外的测试。只要模型的预测结果是这个粗粒度标签下的任何一种子类型,就被判定为预测正确。这种较为宽松的评估指标被称之为 Accuracy (non-leaf)。传统的分类指标(如准确率和F1分数)无法体现错误分类的生物学严重性。例如,将一个“CD4+ T细胞”细胞错分成“CD8+ T细胞”,与错分成完全不相关的“成纤维细胞”,在数值上却被视为同等严重的错误,这显然不符合生物学现实。LCAD 基于细胞本体图计算,其核心是测量目标类别与预测类别之间的层次距离(通过它们的最近公共祖先节点与目标类别节点的路径长度来确定),能够提供更具生物学意义的注释性能评估。
在标准分类指标上,scFoundation 和 UCE 表现最佳。然而,在本体论相关指标上,scCello 表现最优。这一结果与 scCello 基于本体论设计的预训练目标高度一致,证实了该设计能更有效地捕捉和利用细胞类型的层次结构(图 3a)。在跨数据集验证(图 3b)和新细胞类型识别任务(图 3e)中,scFMs 整体展现出更强的泛化能力与OOD(Out-of-Distribution)检测能力。研究人员还通过模型集成实验验证了scFMs之间存在一定的互补性,提示联合使用可能进一步提升综合性能(图 3d)。
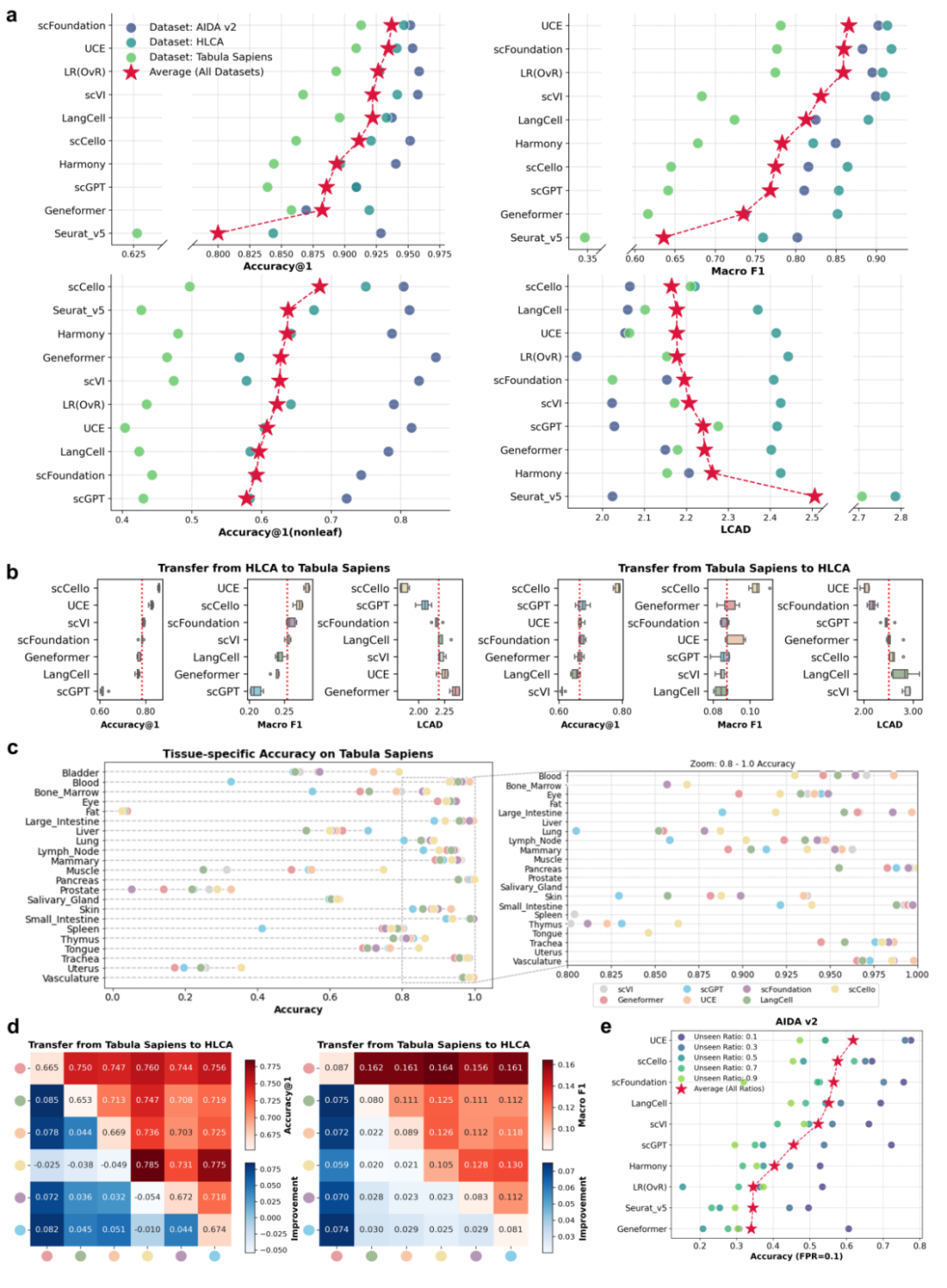
图3. 细胞类型注释实验结果
临床相关任务:scFMs提供了更加平滑的表征空间
本研究进一步探索了scFMs在临床相关任务中的性能优势,包括来自肿瘤患者样本的癌细胞识别和药物敏感性预测。
癌细胞识别任务采用从 TISCH2 数据库收集的来自4种组织的 7 种癌症类型的数据构成,通过留一组织法(leave-one-tissue-out)交叉验证来评估模型的跨组织泛化能力。基线模型采用 Cancer-Finder,通过将模型的输入从原始基因表达替换为 scFMs 提取的细胞嵌入来进行性能比较。scFMs 在此场景下展现出天然的优势,由于它们的输入是可变的,能够保留每个数据集独特的生物信息,避免关键生物学基因的丢失。结果表明,UCE 模型始终优于基线模型,证明了其具有跨组织同质性的细胞表征空间。UCE 受益于其大规模、包含疾病状态的多样化预训练语料库和“基因是否表达”的二元学习目标,可能导致了更加鲁棒的嵌入空间来区分正常细胞和恶性细胞。scGPT 和 scVI 模型在 4 种组织中均取得了具有竞争力的结果,两者都执行 HVGs 选择,突出了 HVGs 在癌症细胞识别任务中的重要作用。此外,scGPT 的优势可能源于其在超过 3300 万个正常细胞上的基于回归的预训练,这建立了一个精确的“健康”转录组的定量基线。
药物敏感性预测任务在4种药物的跨域(bulk → single-cell)数据中进行测试,基线模型为SCAD,通过将模型的输入从原始基因表达替换为 scFMs 提取的细胞嵌入来进行性能比较。结果表明,所有 scFMs均优于基线模型,其中 scFoundation 和 scGPT 表现最佳。scFoundation 的优势可能归因于其测序深度感知(read-depth aware) 的预训练和专门设计的特征提取策略,这使其能够推广到具有不同测序深度的数据,包括 bulk RNA-seq。同样地,scGPT将定量的基因表达值转化成细胞特异性的分箱值,缓解了单细胞数据和bulk数据分布的差异。
为了进一步分析scFMs带来性能提升的内在原因,研究人员通过“粗糙度指数”(Roughness index,ROGI)评估了表征空间的平滑程度,并通过统计分析证明ROGI和分类性能(AUROC)存在显著的负相关性。这些结果表明,scFMs带来的性能提升可以归因于更平滑的表征空间,从而增强了机器学习算法的可建模性,而原始计数数据包含更多数据噪声,带来了更严峻的优化挑战。 这也表明 ROGI 可以评估特定数据集中零样本细胞表征的内在质量,在无需额外训练模型的情况下,为模型选择提供可靠的指导。
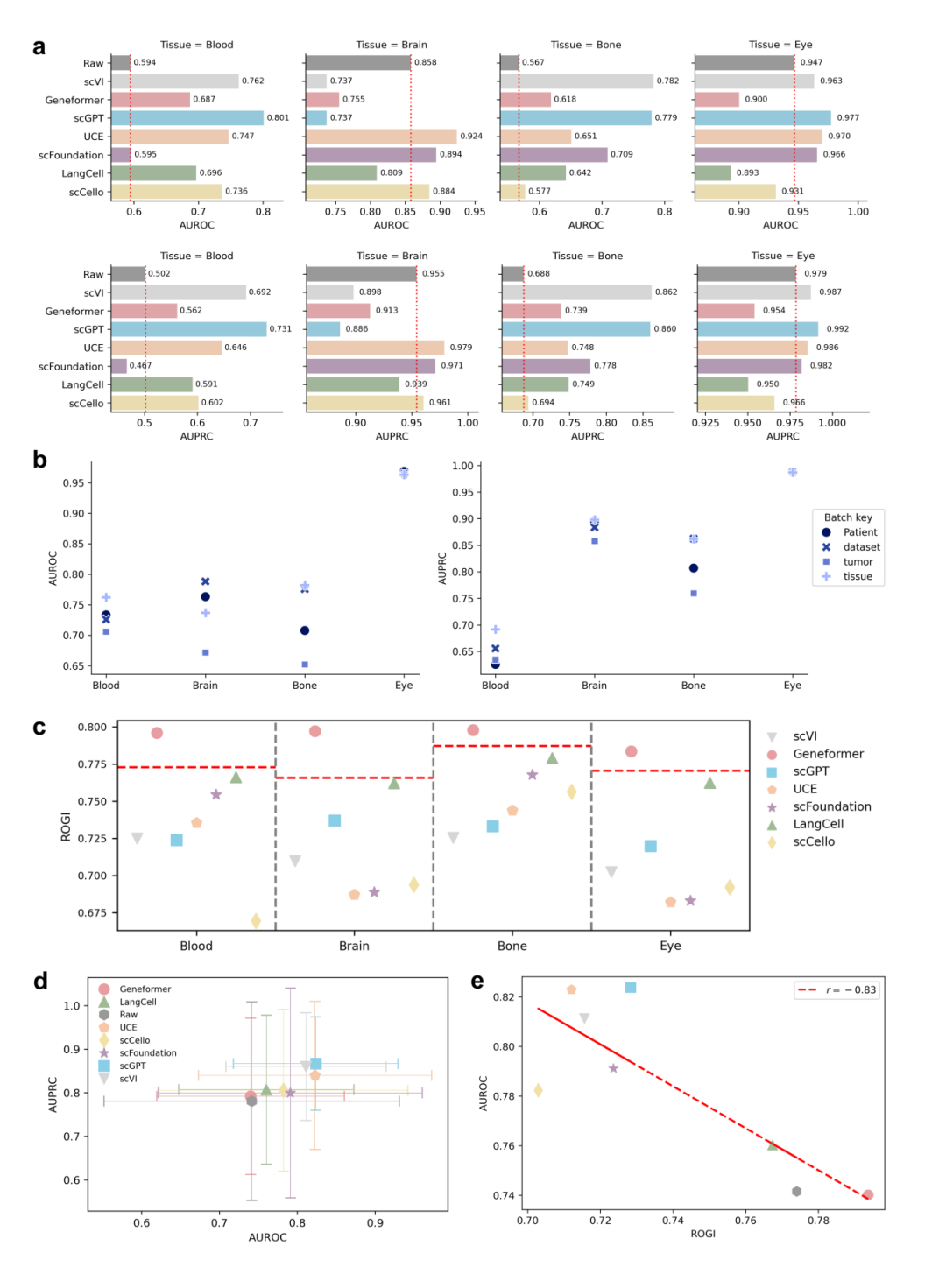
图4. 癌细胞识别实验结果
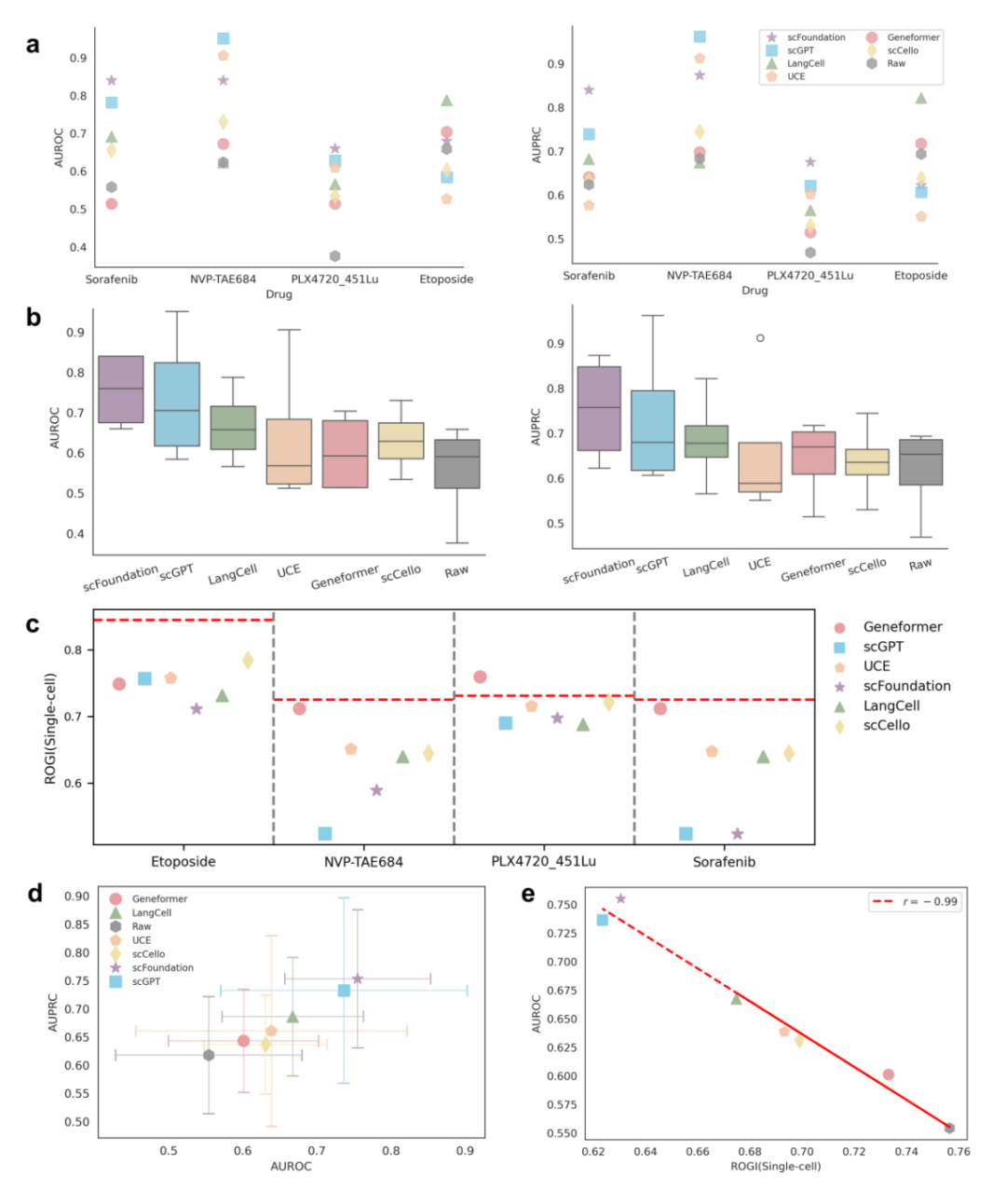
图5. 药物敏感性预测实验结果
3.4 整体性能分析: “没有免费的午餐”
综合上述评测结果,本研究基于非支配排序(Non-dominated Sorting)算法对模型进行了综合排名。该机制避免了人为设置权重,保证用户能够直接从第一层帕累托前沿所代表的最优权衡集合中,更好地选择满足其个性化需求的最佳模型。同时,研究人员基于模型整体性能对三个关键问题进行了解答:
如何评估单细胞基础模型捕捉有意义的生物学见解的能力?
模型的排名随具体指标、数据集、任务和验证场景的变化而变化。因此,基准研究需要更复杂和全面的实验,并使用广泛的可用指标,以充分评估scFMs的泛化能力。本研究计算了 scIB、scGraph-PCA 和 scGraph-OntoRWR 产生的模型排名与其他下游任务排名之间的 Spearman 相关系数。结果表明:(a) 这三个指标产生的排名具有较低的相关性,证实它们提供了正交的视角。(b) 基于scGraph-OntoRWR 的模型排名与其他下游任务排名的平均相关性最高,表明它是衡量模型实际效用更可靠的指标。此外,本体感知分类指标(non-leaf Accuracy和 LCAD)不仅揭示了模型分类性能的细微差异,而还能够更好地指示模型在跨数据集场景下的泛化能力。
在决定使用复杂的基础模型还是简单的机器学习模型时,哪些因素应作为指导?
传统机器学习方法可能在更简单的下游任务上实现最佳性能,而 scFMs 则展现出更好的可重用性和更广泛的场景泛化能力。因此,模型的选择高度依赖于具体应用场景,包括数据集大小、任务复杂度、生物学可解释性需求以及可用的计算资源。scFMs 擅长为大规模数据集提供通用的细胞嵌入空间,而更简单的机器学习模型则更善于在有限的计算资源下高效地适应当前数据集。
是否存在一些基础模型在不同应用场景中始终表现出优越的性能?
本研究的实验结果表明没有单一模型在所有任务中均表现最佳。总体而言,scFoundation 和 UCE 的综合性能最优,它们分别拥有最大的预训练数据和模型参数量。除了规模法则(Scaling law)之外,研究人员还系统地分析了几个影响模型性能的关键设计选择,包括预训练数据、基因表达编码方式、学习目标等,这些因素是解释某些模型在特定任务上表现优异的关键。在具体任务中,ROGI可以作为无需训练即可评估模型潜力的代理指标,联合使用多个scFMs进行模型集成也是潜在的应用方向之一。
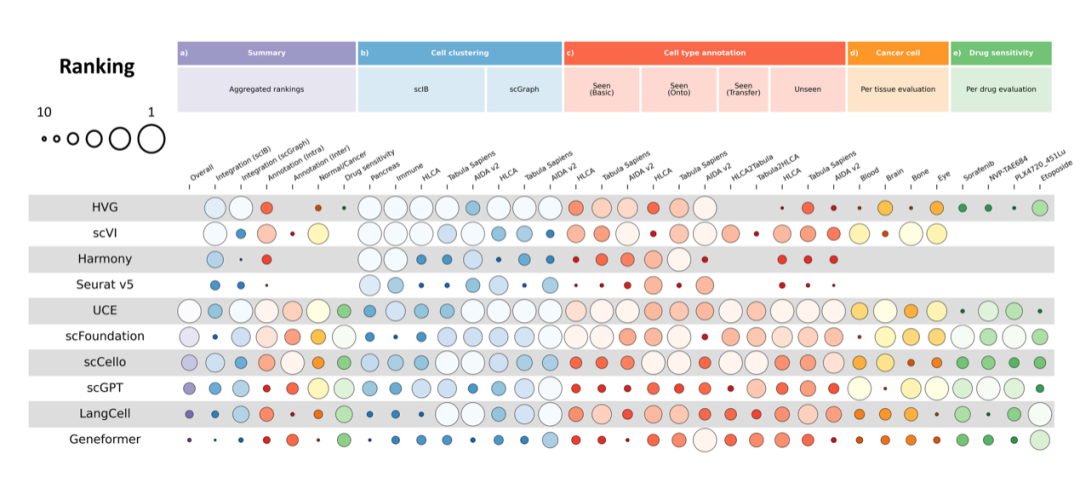
图6. 基于非支配排序算法的总体性能排名
总结
本研究系统评估了单细胞基础模型在多种生物学任务中的零样本能力,提出了基于细胞本体论的评估视角,并提供了面向实际应用的模型选择指南。研究指出,scFMs在构建通用细胞嵌入空间、捕捉生物学结构方面具有显著优势,具有更好的可重用性和场景泛化能力。此外,基于 Transformer 的单细胞基础模型能够提供上下文基因嵌入、细胞嵌入、注意力权重、基因表达预测,甚至能够从头生成 scRNA-seq 数据,显著扩展了它们的应用范围。不可否认的是,在资源受限或任务较为简单时,传统方法仍具竞争力。未来,scFMs的发展方向包括整合多组学数据、为bulk数据和单细胞数据开发统一的细胞嵌入,设计更具生物学意义的预训练任务等。本研究弥补了单细胞基础模型的理论进展与实际应用之间的差距,为单细胞领域的基准测试和方法开发提供了新颖且宝贵的见解。
参考资料
Wu, J., Ye, Q., Wang, Y. et al. Biology-driven insights into the power of single-cell foundation models. Genome Biol 26, 334 (2025).
https://doi.org/10.1186/s13059-025-03781-6
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢