
DRUGONE
准确而快速地评估蛋白–配体结合亲和力是早期药物发现的关键。然而,物理学方法虽准确但昂贵,经验打分函数虽高效却不稳定;现有机器学习方法在泛化到新蛋白或新化学系列时常失效。研究人员提出 CORDIAL:一种带有物理归纳偏置的深度学习框架,通过仅基于相互作用的距离依赖特征来学习蛋白–配体界面的物化规律,避免直接参数化化学结构。在模拟真实应用场景的CATH 超家族留出(LSO)验证中,CORDIAL 在未见蛋白家族上保持了预测性能与概率校准,优于多种当代 3D-CNN 与 GNN 基线,并显著强于非 ML 的打分基线。该结果证明:将任务相关的物理原则编码进模型架构,是获得可泛化结构-基础亲和力排序模型的有效途径。

药物发现需要在庞大化学空间中高效筛选高质量命中分子。传统计算路线在“精度–速度”之间难以兼顾;ML 虽被寄望弥合两者,但广泛暴露出分布外(OOD)泛化差的问题。研究人员指出,主流结构中心表示(体素/图)易在有限数据上学习到与具体子结构相关的伪相关,与可迁移的分子相互作用物理相竞争;再加之常见的验证切分(随机、时间、留一个蛋白、序列相似度)无法真正检验 OOD 能力,导致模型在真实前瞻应用中表现“看上去很美、用起来失灵”。
方法
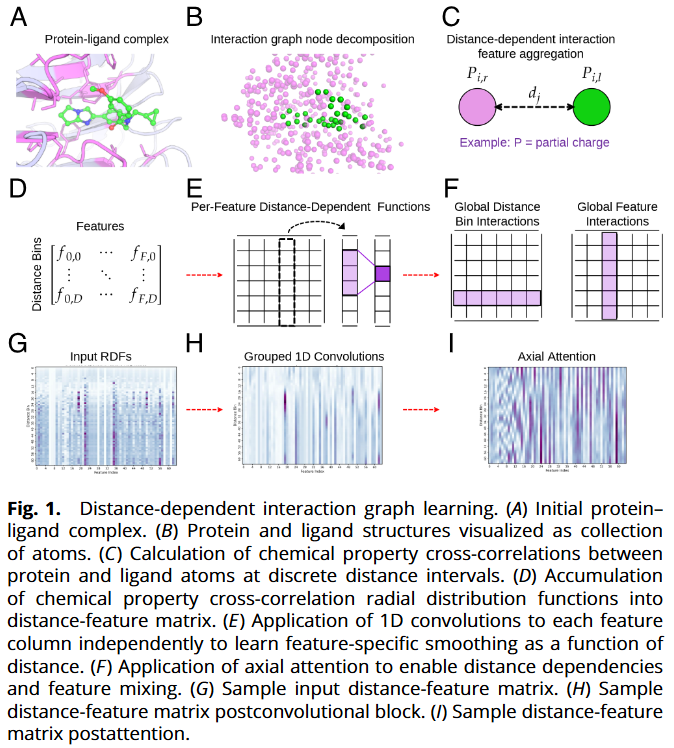
表示与特征:CORDIAL 不直接编码化学拓扑或三维坐标,而是对蛋白-配体原子对在 0–16 Å 距离范围内的物化属性交叉相关进行分箱累计,构成“相互作用径向分布函数(RDF)矩阵”(64×64:距离×特征)。特征覆盖带电/极性、氢键、疏水/芳香、范德华/极化等对偶组合,并区分同号/异号等物理方向性。
模型架构:首先对每个特征通道沿“距离”维做分组一维卷积(学习各类相互作用随距离的平滑/峰值);随后用轴向自注意力先按距离、再按特征进行全局建模,实现远程距离依赖与特征间耦合;最后用 MLP 输出8 个累积阈值(pKd ≥1…≥8)的序等级概率。
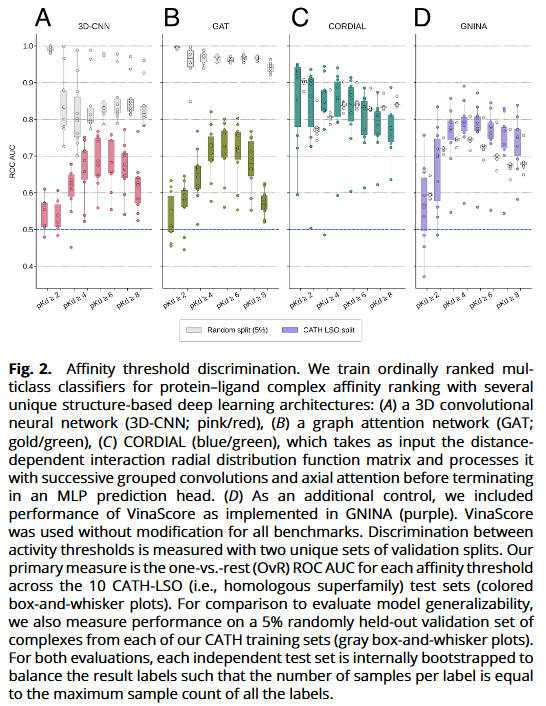
训练与验证:以序等级分类范式训练(每个阈值独立二分类损失),并在随机 5% 验证与CATH-LSO两套切分下评估;与典型 3D-CNN、GAT(图注意力)及传统打分基线对比。
结果
在“未见蛋白家族”上的亲和力阈值判别
在随机验证下,各深度模型表现均佳;但切换至 CATH-LSO 时,3D-CNN 与 GAT 的 ROC-AUC 明显下降,尤其在低阈值段(pKd ≥1–4)与最高阈值段(≥8);而 CORDIAL 的中位 ROC-AUC 基本保持,显著优于非 ML 基线。这表明仅基于相互作用空间的表示与架构抑制了子结构伪相关,提升了 OOD 判别能力。
混淆矩阵与序等级指标
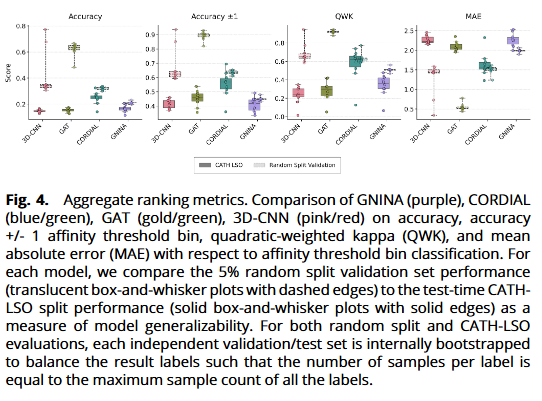
从归一化混淆矩阵看,随机分割下三类深度模型均呈对角主导;但在 CATH-LSO 上,GAT/3D-CNN出现更多偏离对角的误判;而 CORDIAL 在多数 LSO 组上仍保持更清晰的对角带。
量化指标方面:在 CATH-LSO 下,GAT/3D-CNN 的 QWK 下探至 ~0.2–0.3,MAE 上升;CORDIAL 维持中位 QWK ~0.65、MAE ~1.5 个等级,且 **“±1 等级准确率”**最高,体现更稳健的序等级排序能力。

按蛋白目标的细粒度评估
研究人员对 10 个代表性目标(各来自一个 LSO 测试集)进一步作图:GAT/3D-CNN在多目标上给出分散预测;而 CORDIAL 更接近对角集中。也有全体方法均困难的个例(如某酶家族成员),提示特定超家族的理化特性对单点亲和力模型构成挑战。

模型概率校准(OOD 条件)
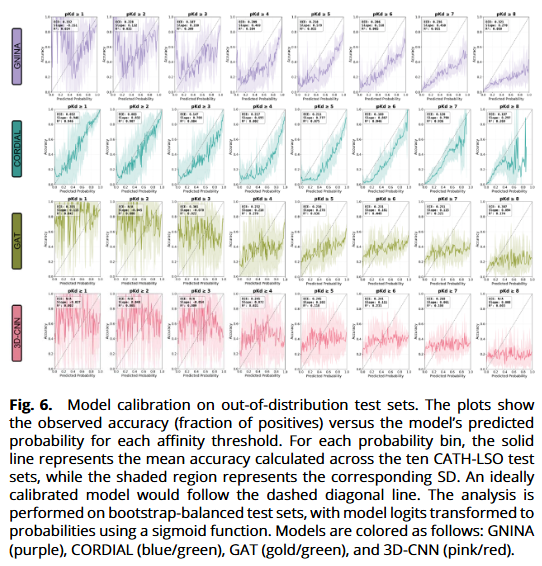
在 CATH-LSO 上,传统打分在低阈值区间校准差;GAT/3D-CNN的概率–准确率曲线近乎扁平,难以作为真实概率解读。CORDIAL 在 pKd ≥1–6 的阈值段呈接近对角线的线性关系,仅在更高阈值(≥7, ≥8)出现过置信/压缩现象,但总体仍显著优于对照。这与其按阈值独立学习的训练策略及物理归纳偏置一致。
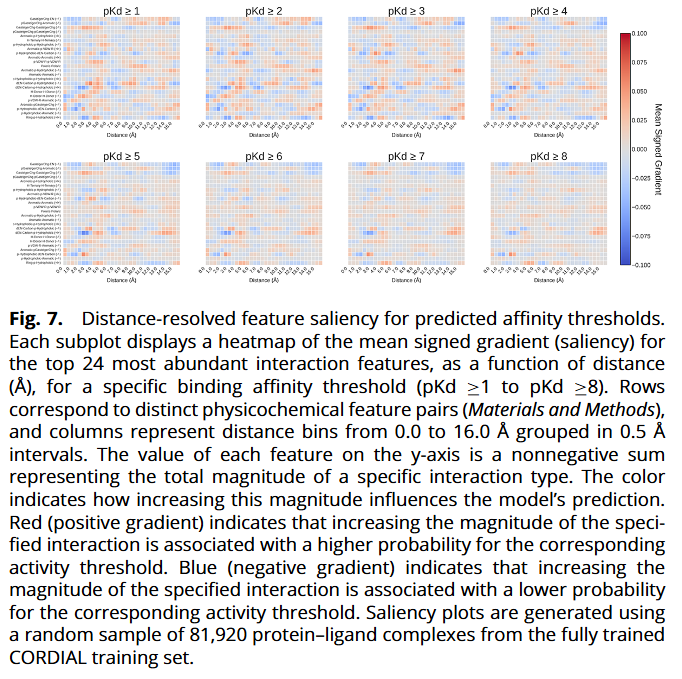
特征显著性与物理可解释性
研究人员对 CORDIAL 做显著性(梯度)分析:
在最低阈值,模型学到短距离强接触普遍惩罚(如 <~2.5 Å 的排斥)。
随着阈值提升至中高区间,在物理合理距离(~2.5–5.0 Å)上,若干相互作用(如带电/氢键/芳香-疏水耦合等)出现连续的正向贡献带;
最高阈值下显著性图更“稀疏”,符合高亲和力依赖少数优化相互作用的直觉。
整体呈现出距离依赖、化学合理、随阈值层级演化的解释模式。
讨论
研究人员通过严格的 CATH-LSO 基准与交互作用-唯一路线的架构设计,验证了如下结论:
架构归纳偏置很关键。结构中心 3D-CNN/GNN 的灵活性高,但在有限数据下易学到与子结构频次相关的捷径;而相互作用-唯一表示抑制伪相关、更易学习可迁移的物理规律,因此在 OOD 评测更稳健。
良好校准有实际价值。在前瞻筛选中,分数若可作为真实活动概率解读,将减少“逐靶点再校准”的成本,提升从虚拟筛选到实验验证的衔接效率。
限制与展望。现版本牺牲了部分几何分辨率(采用 1D 距离剖面)且未显式处理构象/姿势判别;未来可引入几何关系约束、可学习的原子对嵌入、更细致的数据清洗与靶标特异的药效团增强。同时,基准还可更严格地处理多结构域蛋白的潜在泄漏,推动形成类似结构预测领域那样的“社区级挑战”。
总体而言,CORDIAL 提供了一条可落地的路径:在适度规模数据上,通过将任务物理显式融入表示与架构,获得可泛化、可解释、校准良好的结构-基础亲和力排序模型;可与传统物理方法互补,加速早期命中发现与可信 AI 工具的构建。
整理 | DrugOne团队
参考资料
B.P. Brown, A generalizable deep learning framework for structure-based protein–ligand affinity ranking, Proc. Natl. Acad. Sci. U.S.A. 122 (42) e2508998122,
https://doi.org/10.1073/pnas.2508998122 (2025).

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢