
在单细胞水平上测定蛋白丰度,有助于以高分辨率理解细胞过程和疾病进展中的生物学机制。然而,现有单细胞蛋白质组学技术面临覆盖度有限、通量受限、灵敏度不足、批次效应明显、成本高昂及实验要求严格等问题。受自然语言处理中的“翻译”机制及遗传学中心法则启发,研究人员提出了一种名为 scTranslator 的预训练大型生成模型,可基于单细胞转录组推断缺失的单细胞蛋白质组信息,从而生成多组学数据。系统评估表明,scTranslator 在多种测序技术(CITE-seq、REAP-seq、NEAT-seq、空间CITE-seq等)、多种细胞类型(单核细胞、巨噬细胞、T细胞、B细胞)、不同组织(血液、肺、脑)及多种疾病背景(感染性、代谢性、肿瘤性疾病)中均具有出色的准确性、稳定性和泛化能力。此外,scTranslator 在多个下游任务中表现优异,包括基因/蛋白互作推断、扰动预测、细胞聚类、批次校正及癌症数据中细胞来源识别,显示出其在单细胞多组学研究和疾病机制解析中的广泛潜力。

自单细胞转录组测序技术(scRNA-seq)问世以来,生物学研究进入了单细胞时代。scRNA-seq 揭示了不同系统中细胞的转录图谱,但其结果作为蛋白水平的代理仍受限,仅有约40%–60%的蛋白丰度变化可由 mRNA 水平解释。在单细胞尺度上,这一相关性因细胞间的转录状态、翻译速率及蛋白降解速率差异而进一步减弱。
然而,蛋白作为细胞功能的直接执行者,对于解析细胞分化、命运决定、信号传导与疾病进展至关重要。受限的单细胞蛋白质组技术阻碍了对应数据的生成与分析。一个可行途径是基于大规模 RNA–蛋白数据,通过计算模型预测蛋白水平。
近年来,大规模预训练模型在自然语言处理与计算机视觉中表现出卓越的生成与理解能力,促进了科学研究的跨领域应用。传统的机器学习方法依赖手工特征工程,容易受到数据稀疏和泛化性不足的限制,因此,探索预训练大型生成模型在单细胞多组学中的潜力具有重要意义。
研究人员基于语言翻译与中心法则的类比提出 scTranslator,其将单细胞转录组“翻译”为蛋白质组。通过对超过18000例患者和240万细胞的配对RNA–蛋白数据进行两阶段预训练,模型可从转录数据中生成相应的蛋白丰度。scTranslator 采用改进的 Transformer 架构,结合重新索引的基因位置编码(GPE)、快速注意力机制 FAVOR+ 以及非自回归式解码策略,实现高效、可扩展、可解释的蛋白预测。
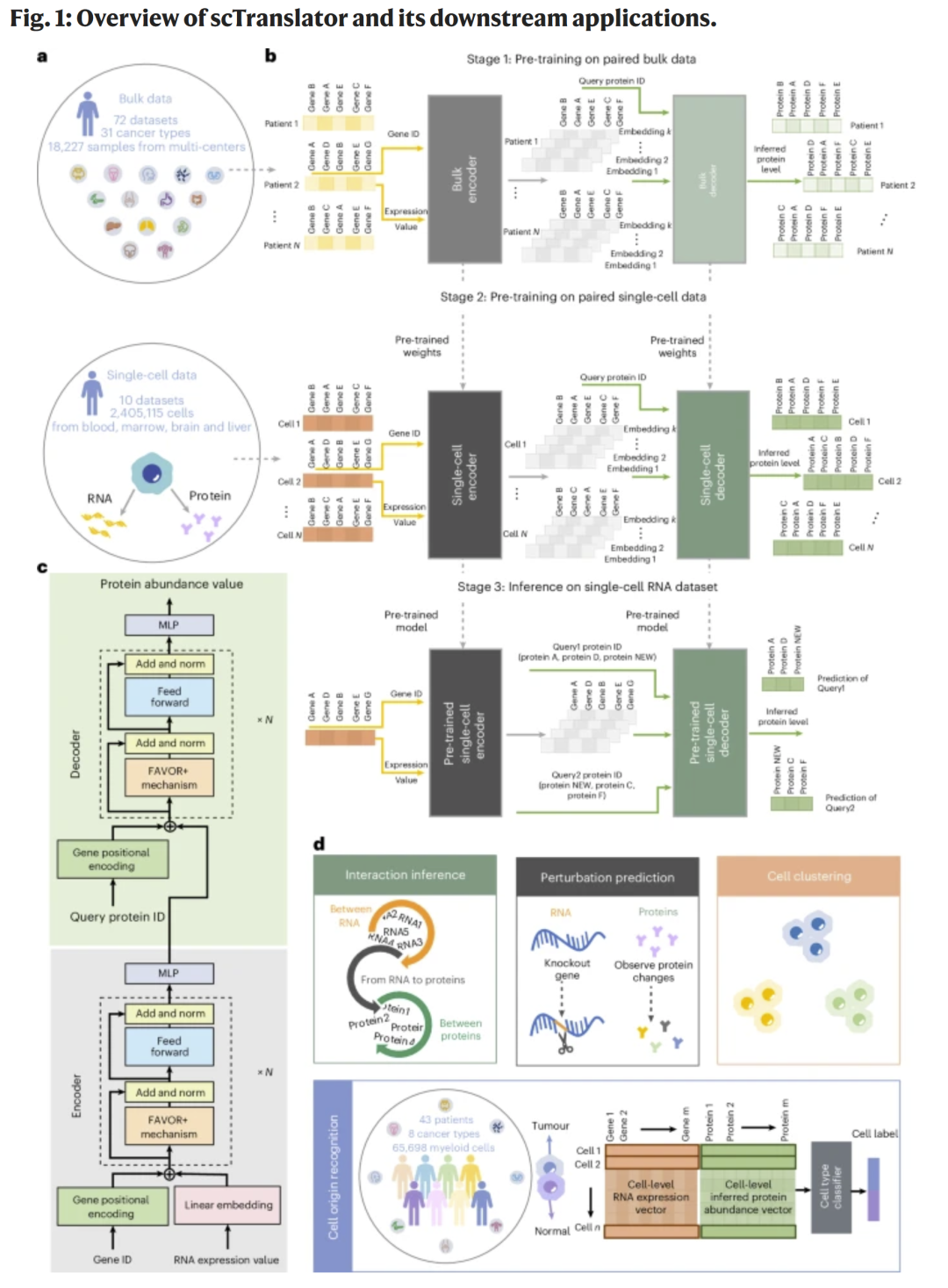
图1|scTranslator框架概述与下游应用
方法概述
研究人员构建了三阶段框架:
阶段一: 在配对的体细胞级(bulk)RNA–蛋白数据上进行预训练,学习广泛的分子规律。
阶段二: 在配对的单细胞数据上进一步预训练,细化模型以捕捉细胞间差异。
阶段三: 在下游任务中利用已预训练模型推断蛋白丰度,可选择是否进行微调。
模型采用编码器–解码器结构。输入的基因表达值经嵌入层映射到向量空间,并加入 GPE 位置编码以区分不同基因。通过 FAVOR+ 注意力机制捕捉长程依赖关系。解码器部分根据蛋白 ID 生成预测丰度,非自回归设计显著提升了推断效率。该框架在推断蛋白丰度后,可支持多种下游任务,包括互作推断、扰动模拟、聚类与细胞来源识别。
结果
scTranslator 在体细胞与单细胞数据上均具有高精度预测能力
两阶段预训练后,模型在训练集和测试集上的表现一致,泛化性良好。对于 bulk 数据,余弦相似度超过0.98;对于单细胞数据,超过0.87。模型能准确预测蛋白及其剪接异构体(如CD45家族),并在多数蛋白上达到高相关性。约50%的蛋白预测相关度超过0.7,富集分析显示高可预测蛋白多涉及信号受体与转导活动。消融实验表明,双阶段预训练显著优于仅在 bulk 或未预训练模型,强调了预训练的重要性。
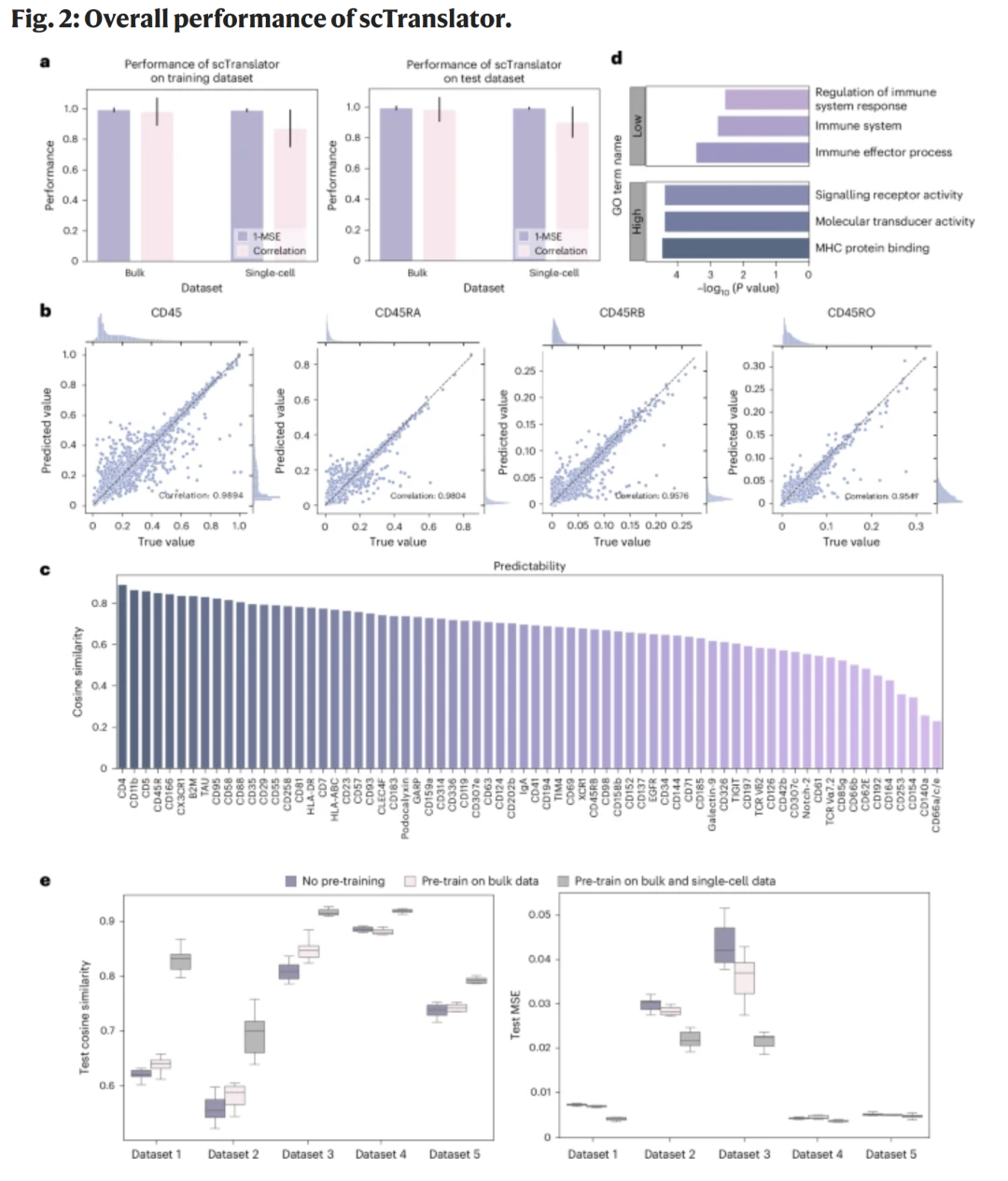
图2|scTranslator在体细胞与单细胞数据上的整体性能评估
与现有方法的系统性基准对比
研究人员将 scTranslator 与九种单细胞多组学或RNA–蛋白预测框架(如 cTP-net、sciPENN、Seurat、BABEL 等)进行比较。结果显示,在少样本(few-shot)实验中,scTranslator 在五个数据集上均显著优于所有对照方法,在余弦相似度、皮尔森相关系数及误差指标上均居首位。即便仅用20个细胞微调,模型仍能达到使用90%数据训练时的80–95%性能。在不匹配(unaligned)实验中,scTranslator 通过 GPE 模块保持高鲁棒性,展现对未见基因/蛋白的迁移能力。
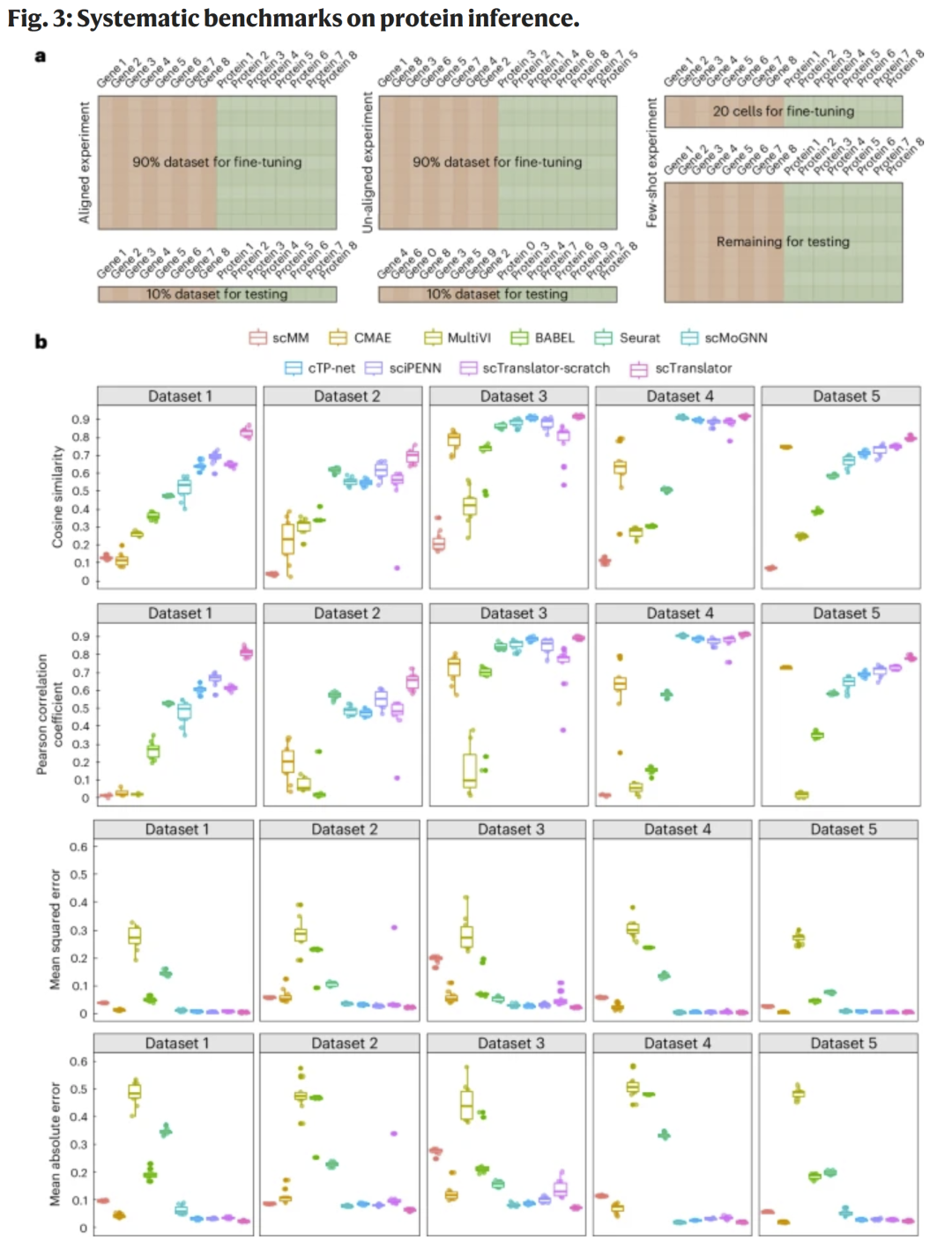
图3|蛋白推断任务的系统性基准比较
在独立数据中的鲁棒表现
研究人员在四个独立数据集(ECCITE-seq PBMCs、CITE-seq mouse、空间CITE-seq、NEAT-seq)上进行留一类型验证。
ECCITE-seq PBMCs: scTranslator 在细胞类型、批次与时间维度上的余弦相似度均高于0.7。
CITE-seq mouse: 尽管预训练仅基于人类数据,模型在鼠源数据上依然保持优异表现,表明跨物种泛化能力。
空间CITE-seq: 模型在不同组织(胸腺、脾脏、扁桃体、皮肤)中准确度最高,显示其从单细胞数据到空间组学的迁移潜力。
NEAT-seq: 预训练模型在核蛋白预测上优于未预训练版本,验证了bulk数据对胞内蛋白学习的有效性。
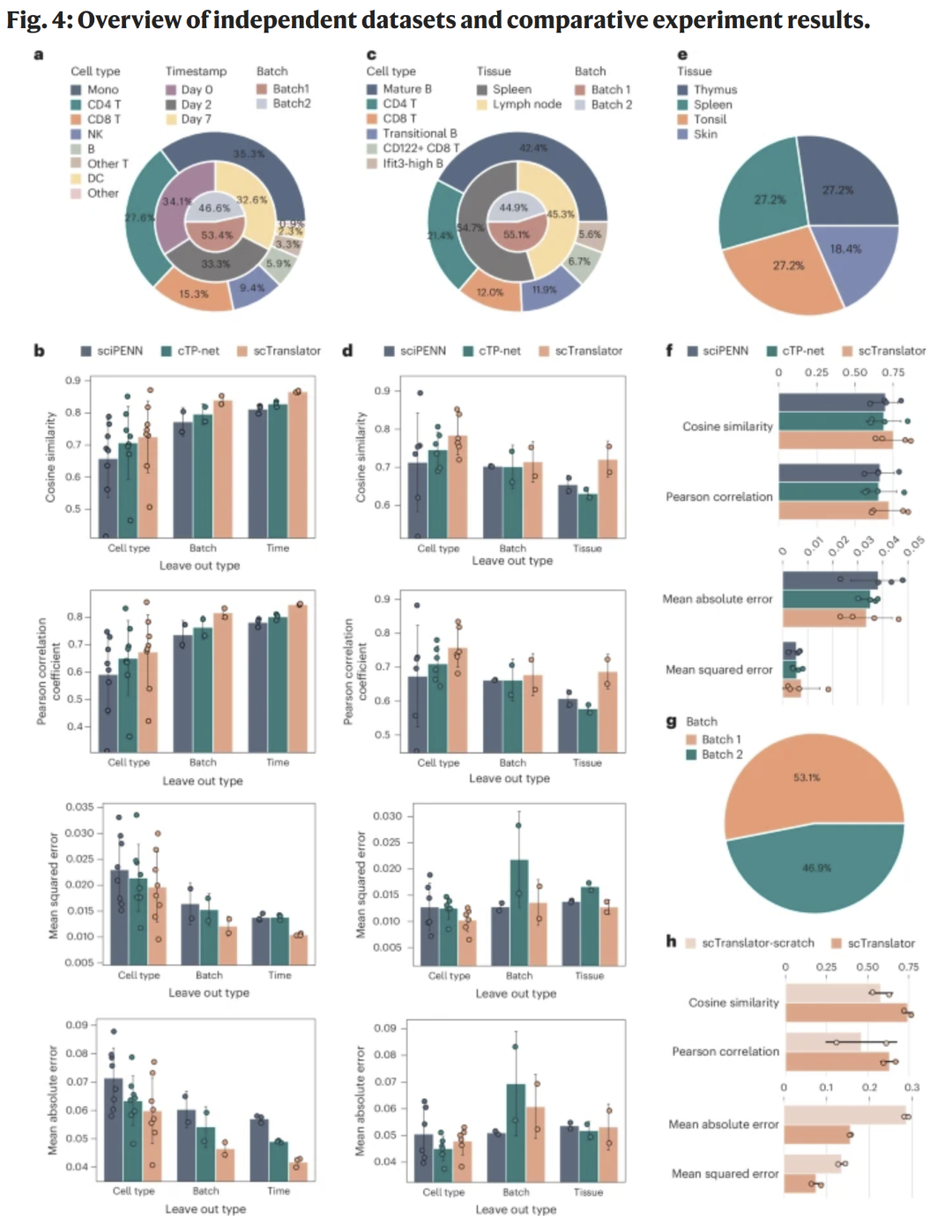
图4|独立数据集上的模型鲁棒性与跨模态泛化性能
基于注意力机制的调控网络与蛋白互作推断
研究人员利用注意力矩阵解析基因–蛋白、基因–基因及蛋白–蛋白关系。高注意力得分的基因多与细胞反应、信号调控相关。可视化结果揭示了STAT3、IRF4、FOXP3等转录因子的靶基因网络,与文献报道高度一致。模型预测的蛋白互作(如CD45RA、CD8a、CD273等)与 STRING 数据库验证结果高度重合,证实其可靠性。
此外,通过输入扰动(如STAT1、JAK1/2、IFNGR1/2敲除)后的RNA表达,模型成功预测蛋白水平变化,与真实Perturb-CITE-seq实验结果高度一致(余弦相似度>0.8),显著减少了实验成本与时间。
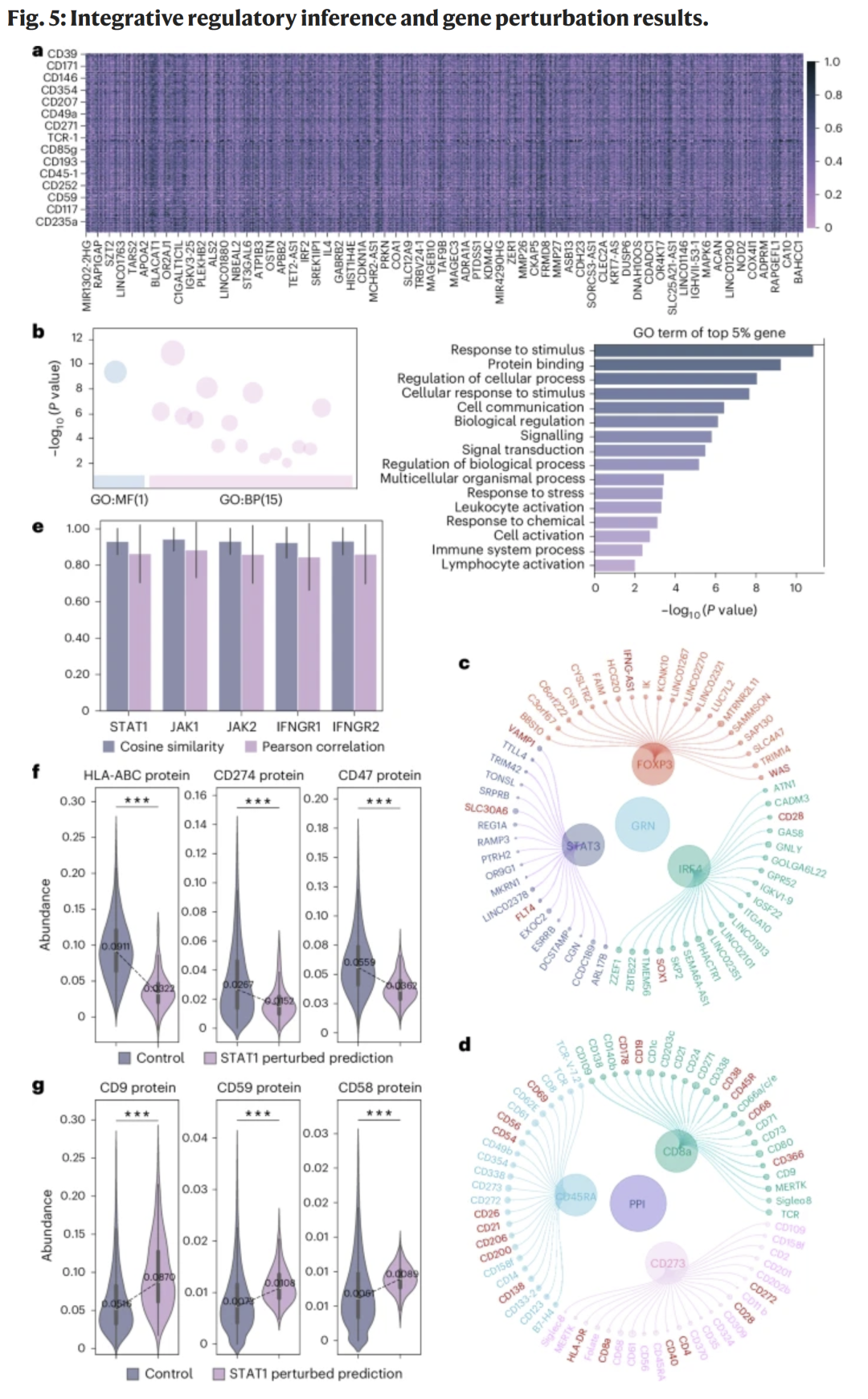
图5|整合调控推断与基因扰动模拟结果
伪蛋白定量促进下游单细胞分析
将scTranslator应用于16000个人PBMCs数据,推断得到的伪蛋白丰度在UMAP空间中呈现清晰的细胞聚类,与真实蛋白结果一致,且明显优于RNA信号的分辨率。模型能有效减少批次效应并保持细胞异质性。研究人员进一步发现新的潜在细胞标记物,为多组学整合分析提供了新的视角。
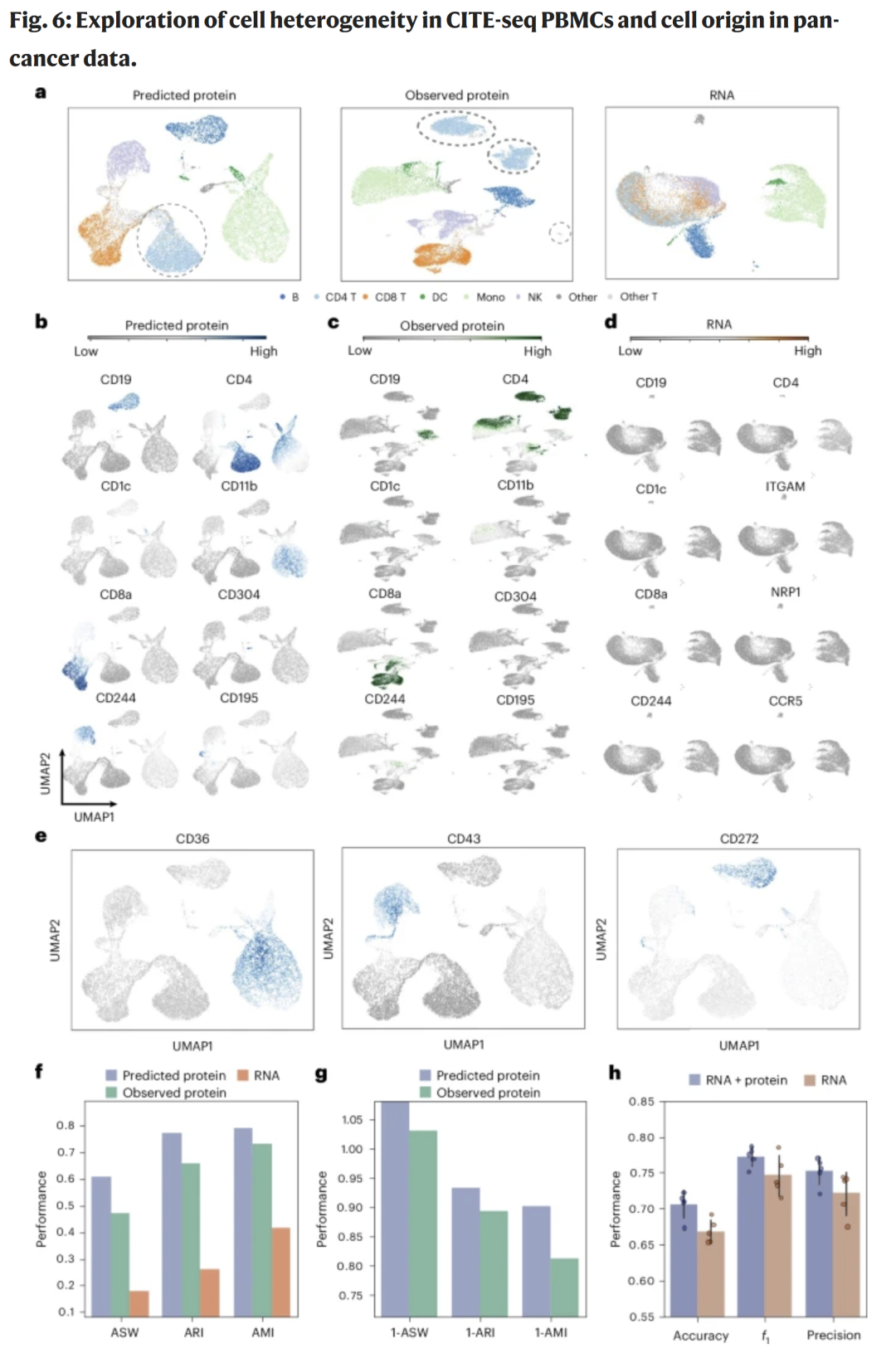
图6|伪蛋白定量在单细胞聚类与批次校正中的应用
讨论
scTranslator 作为首个大规模预训练的单细胞转录组到蛋白质组生成模型,在预测精度、跨模态泛化及生物可解释性上均表现卓越。模型的 re-indexed GPE 模块与 FAVOR+ 注意力机制共同确保其可扩展性与对未知蛋白的外推能力。
研究人员指出,scTranslator 不仅能模拟RNA–蛋白间的非线性关系,还能通过注意力权重揭示基因调控网络与蛋白互作模式,具有强大的知识挖掘潜能。其在细胞聚类、扰动预测及多组学数据补全等任务中的表现,展示了该模型作为“多组学生成引擎”的潜力。
未来,研究人员计划将该框架拓展到代谢组、表观组等更广泛的多组学数据中,促进生物系统层面的整合理解,并加速单细胞生物学与人工智能的融合发展。
整理 | DrugOne团队
参考资料
Liu, L., Li, W., Wang, F. et al. A pre-trained large generative model for translating single-cell transcriptomes to proteomes. Nat. Biomed. Eng (2025).
https://doi.org/10.1038/s41551-025-01528-z

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢