
DRUGONE
不对称加氢是最重要的手性构建反应之一,但现有机器学习模型通常仅适用于单一前手性位点,难以跨底物–催化剂体系泛化,且无法同时预测双前手性烯烃的立体选择性与绝对构型。研究人员提出 ChemAHNet,一个基于不对称加氢机理构建的深度学习模型,通过三个结构感知模块,从 SMILES 输入中自动捕捉原子级空间与电子相互作用。该模型能够准确预测多类催化剂与底物体系中的主要对映体绝对构型,同时通过催化剂–烯烃的相互作用模式计算立体选择性,避免传统 R/S 标记的歧义。ChemAHNet 无需量子化学描述符,具有良好的外推能力,并可推广至其他不对称催化反应,为面向目标的分子设计提供多尺度结构特征洞察。

随着人工智能在化学合成设计中的迅速发展,反应预测模型展现出广泛潜力。然而,大多数模型忽略了关键的立体化学细节,尤其在需要同时预测对映体比例和绝对构型的烯烃不对称加氢中更为突出。此类反应不仅受底物和催化剂结构影响,还涉及立体拥挤、电子效应与双前手性位点识别等挑战。传统模型依赖预定义量化描述符或特定体系训练,在跨底物、跨催化剂的泛化能力不足,难以服务更实际的化学空间探索。因此,研究人员希望借助反应机理本身,在模型中引入“相互作用模式”概念,以实现更准确、更通用的预测系统。
方法
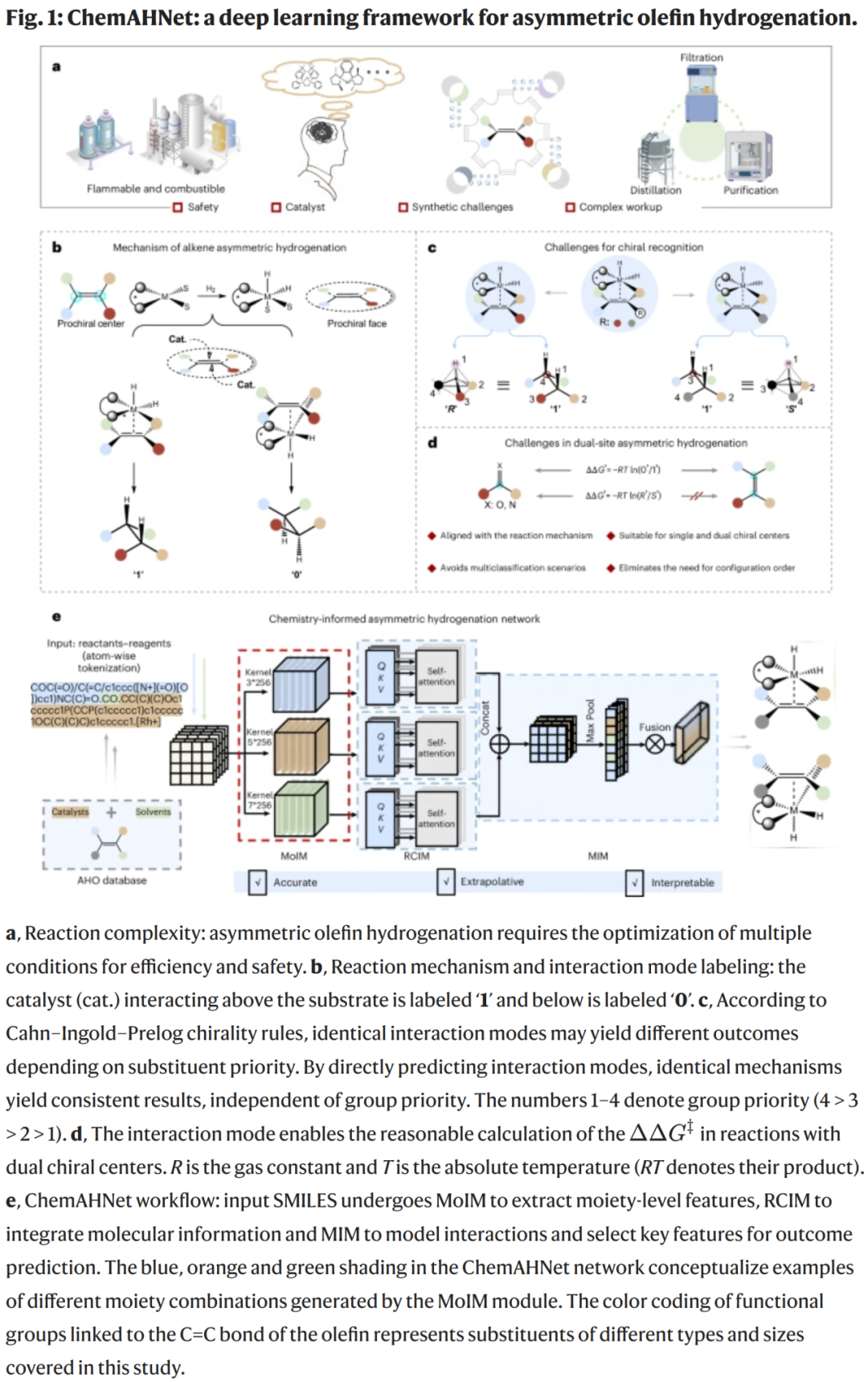
ChemAHNet 包含三个模块:
MoIM(结构单元识别模块):从 SMILES 中提取多尺度取代基与关键结构片段;
RCIM(反应组分整合模块):利用注意力机制整合底物、催化剂与配体的结构信息;
MIM(分子相互作用模块):识别催化剂–烯烃相互作用模式,并同时完成立体选择性的回归预测和绝对构型分类。
该设计遵循烯烃不对称加氢的实际机理,通过对“催化剂位于双键上方或下方”的交互模式建模,使模型自然区分构型路径,避免传统 R/S 标记带来的歧义。
结果
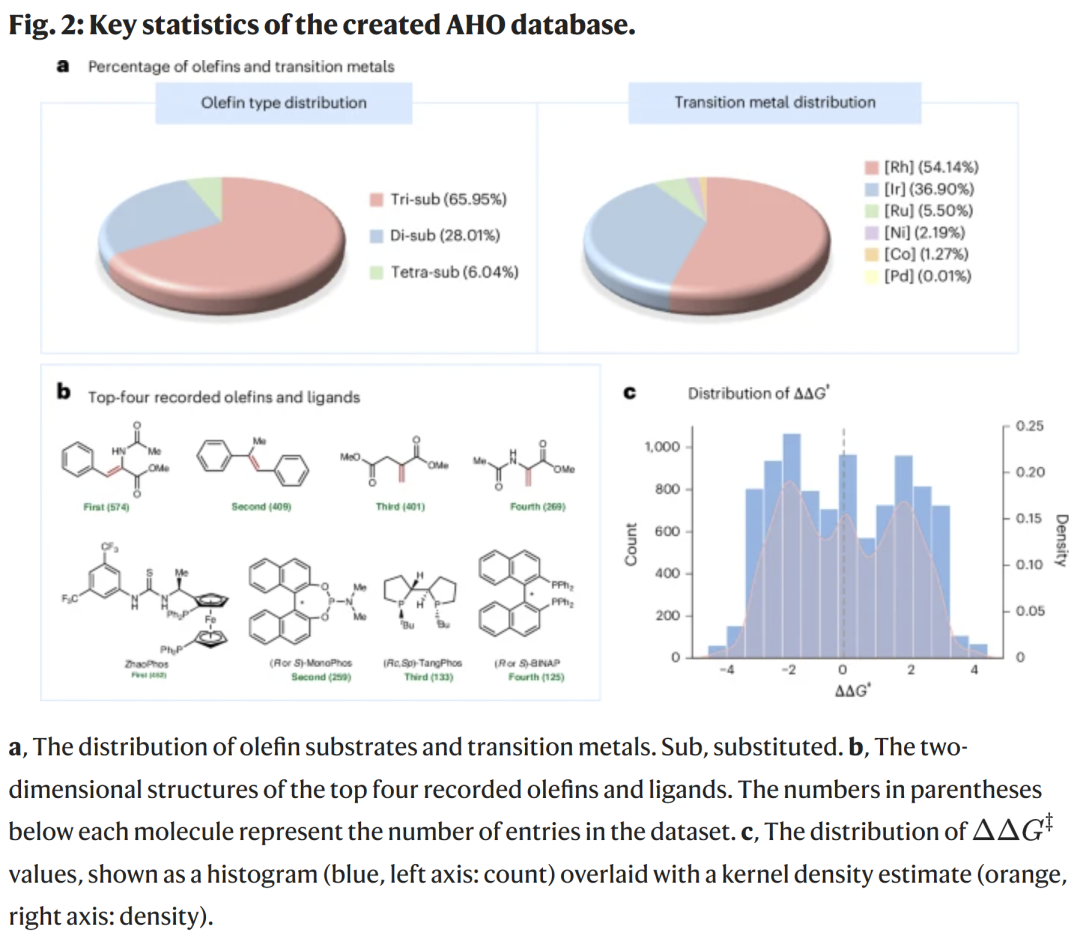
数据集构建与分布特征
研究人员构建了规模为 9,478 条反应的数据集,覆盖三取代、二取代与四取代烯烃,以及多种过渡金属体系(Rh、Ir、Ru、Ni、Co 等)。
立体选择性(ΔΔG‡)分布均匀,有利于模型训练。
数据呈现广泛的结构多样性,适合作为通用预测模型的基础。
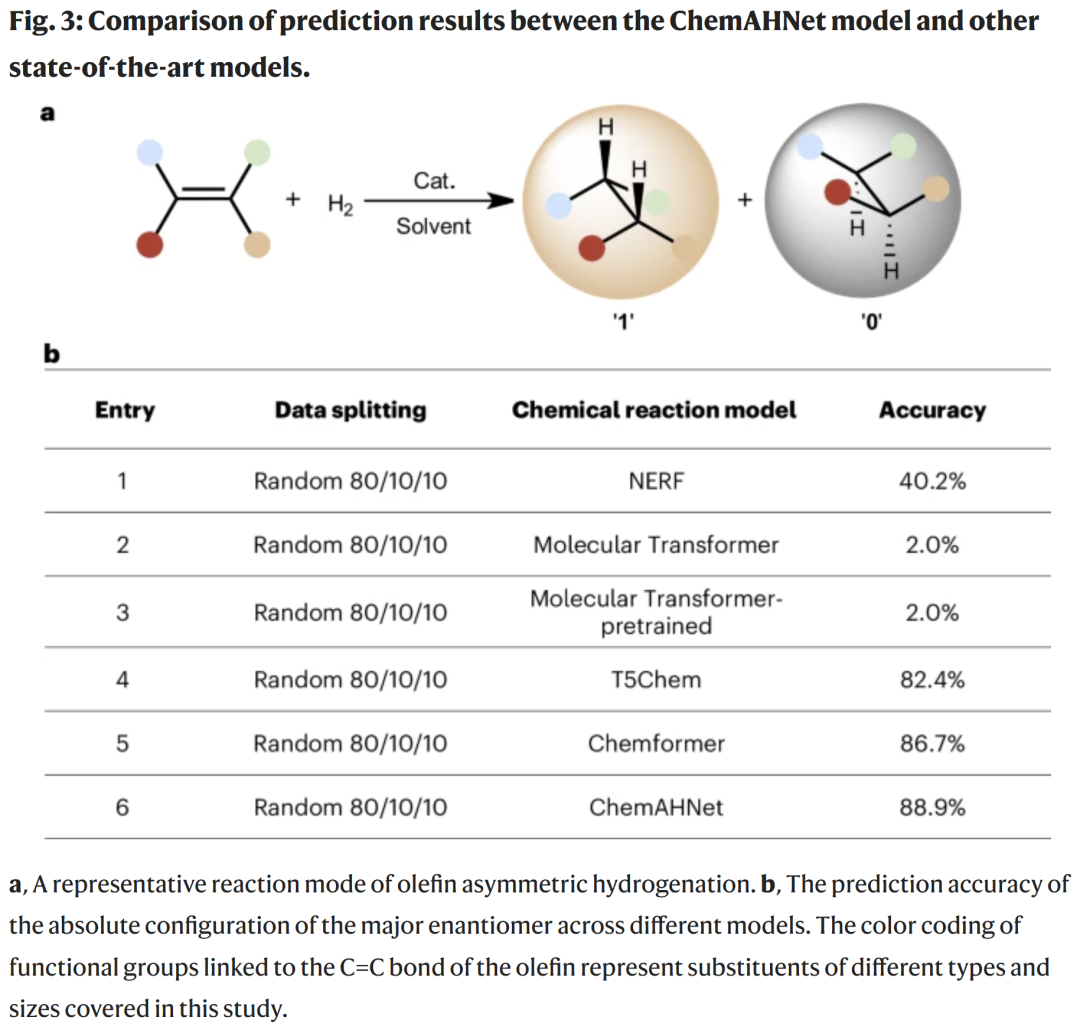
模型整体性能:主要对映体预测
将 ChemAHNet 与多种先进模型对比:
Transformer 类模型在该任务上准确率极低;
预训练模型(T5Chem、Chemformer)达到 82–87%;
ChemAHNet 以 88.9% 的准确率取得最佳结果。
其优势来自对“催化剂–烯烃交互模式”的直接学习,而非依赖可能自相矛盾的 R/S 标签。
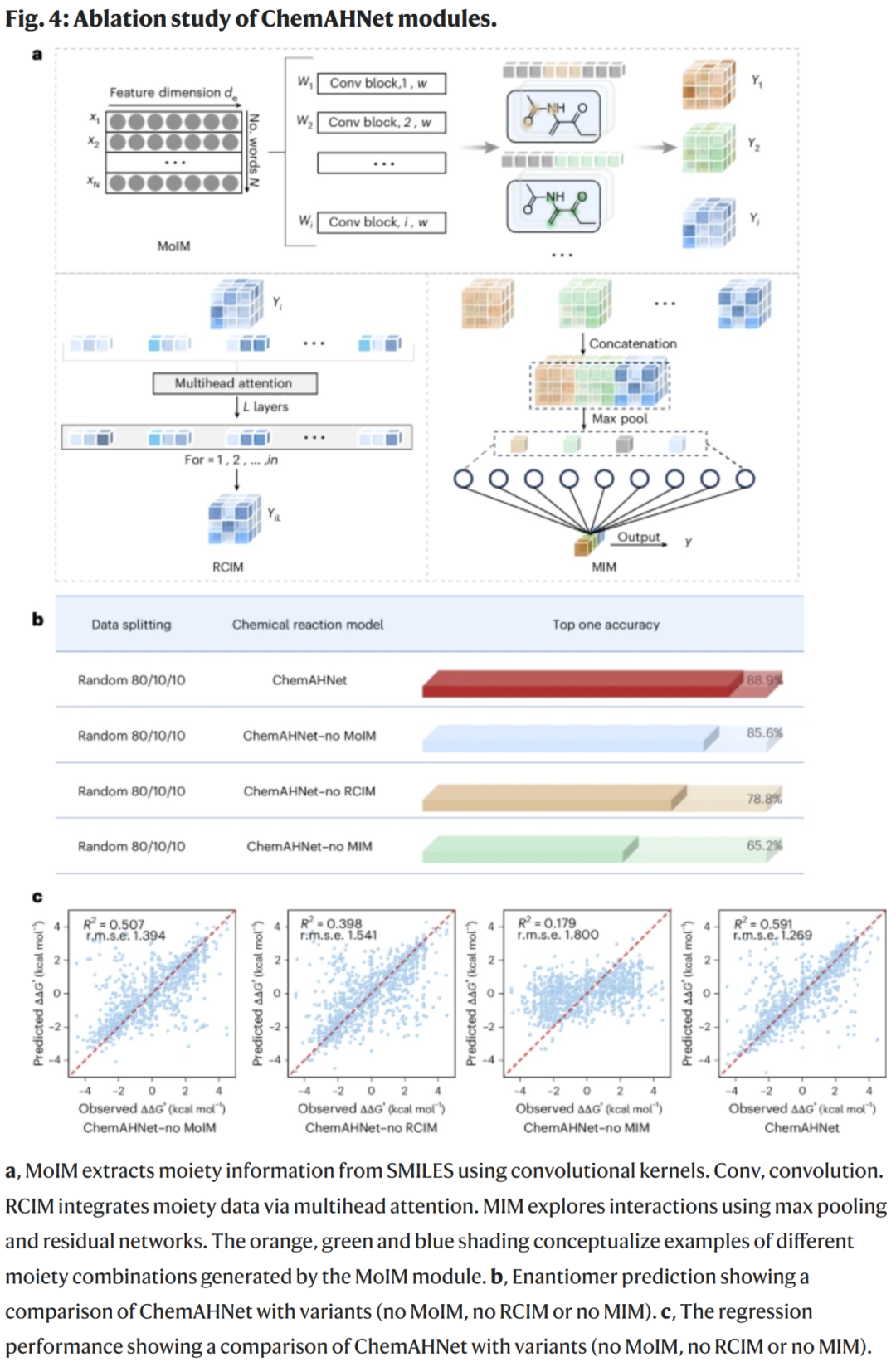
消融实验:模块贡献分析
移除任一模块都会显著降低性能:
删除 MoIM:准确率降低 3.3%;
删除 RCIM:下降 10.1%;
删除 MIM:下降 23.7%,表明相互作用建模最关键。
在立体选择性回归中:
完整模型 R² = 0.591;
移除模块后 R² 显著下降,尤其是 MIM。
立体选择性与绝对构型的联合预测
ChemAHNet 同时预测:
对映体比例(ΔΔG‡)
绝对构型(基于相互作用模式推断)
在特定的 Rh/BINOL 系统中,模型达到:
R² = 0.797
RMSE = 0.41 kcal/mol
优于多种基于图模型与指纹特征的方法。
模型在外延预测中也表现稳定,包括:
跨配体预测
跨金属预测(如在 Ru、Ni、Co 上仍具 78–84% 分类准确率)
高 e.e. 区间预测
跨反应类型泛化能力验证
ChemAHNet 不依赖量化描述符,因此可轻松迁移到其他不对称催化反应。
在以下体系中均取得高性能:
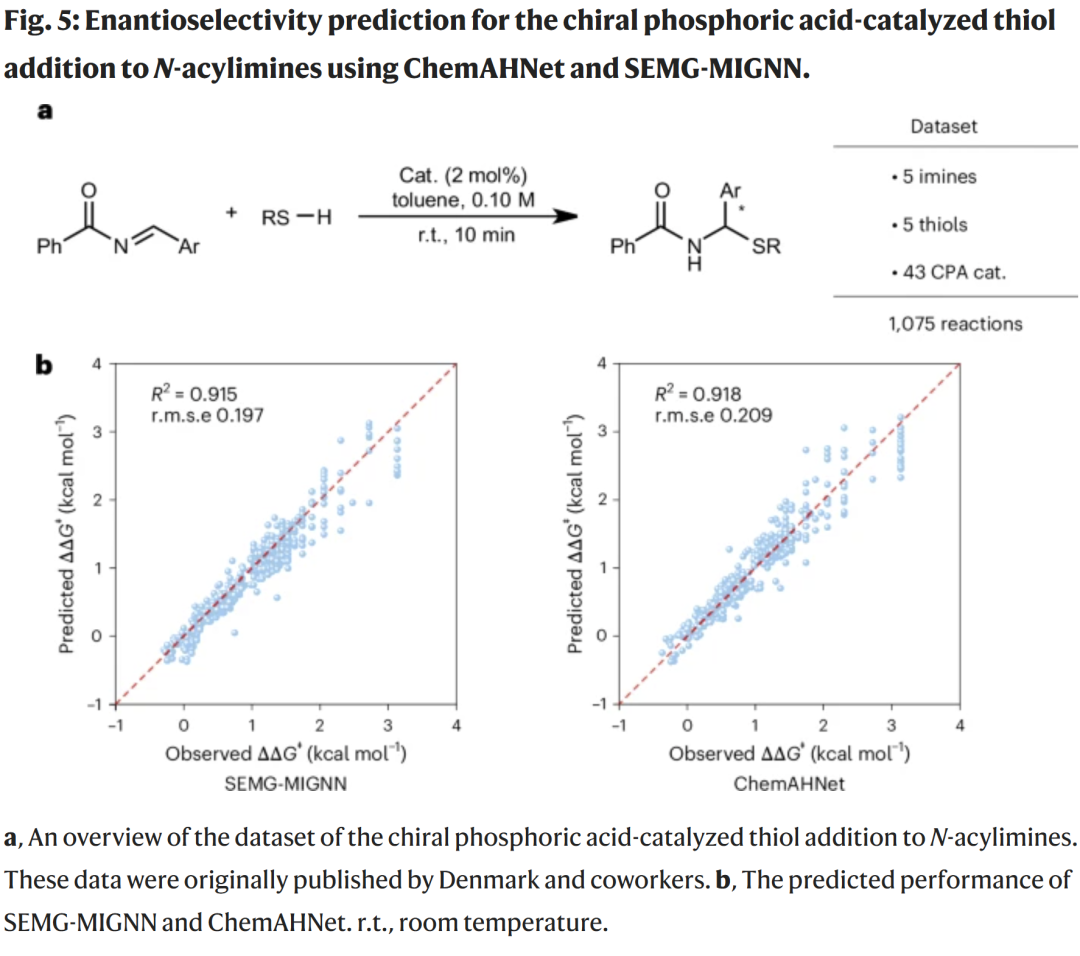
CPA 催化硫醇加成反应(R² 约 0.92,与最佳模型相当甚至略优)
其他三类不对称催化反应(R² = 0.73–0.85)
说明 ChemAHNet 具有跨机理的强泛化能力。
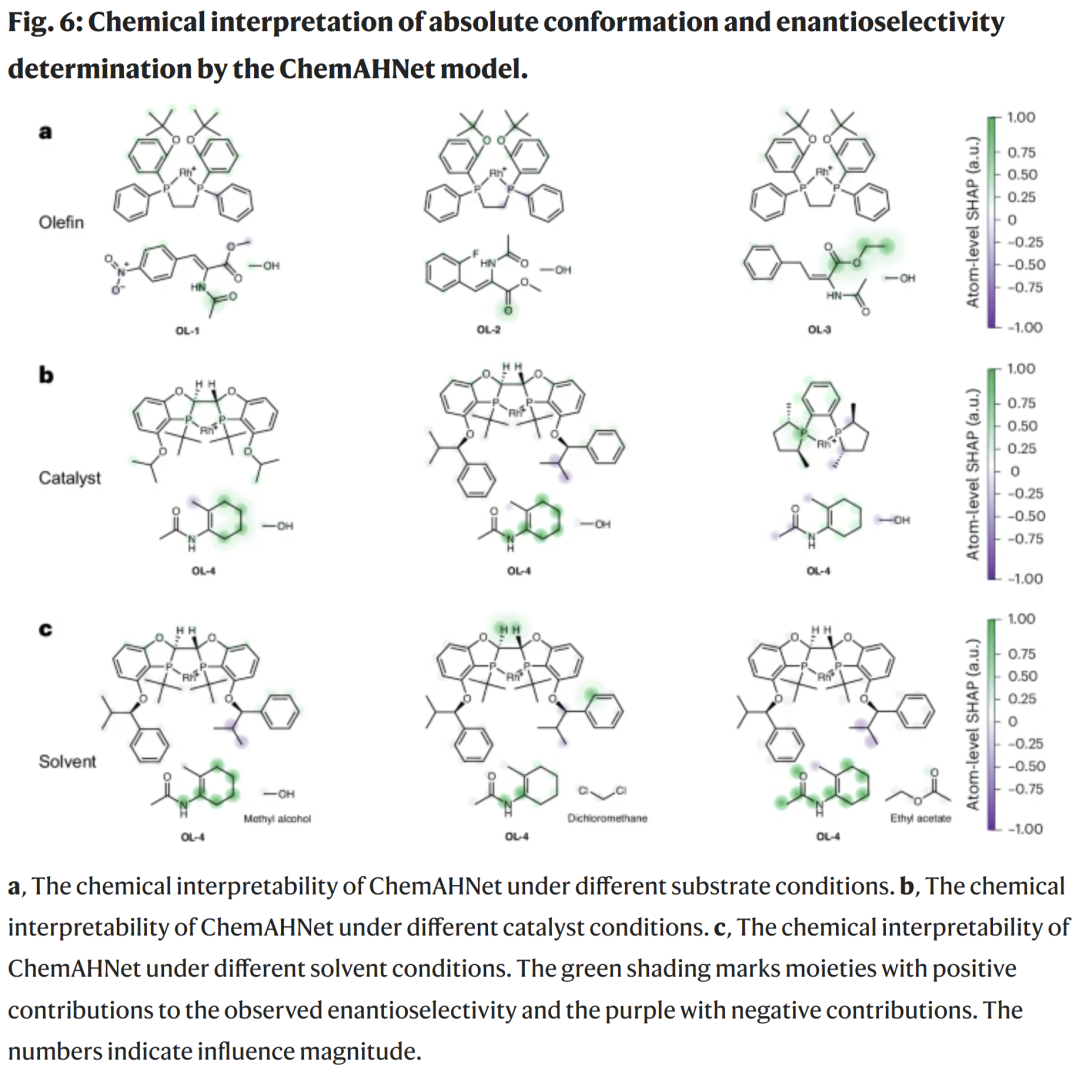
模型可解释性分析
借助 SHAP + XSMILES,模型可定位影响立体选择性的关键原子或取代基。
示例:
酯基、氨基等具有电子吸引/给电子能力的基团被模型赋予更高权重;
不同催化剂下,模型能识别底物环结构的立体阻碍;
不同溶剂的影响较小,与实验一致。
讨论
ChemAHNet 展示了化学机理与深度学习结合的优势:
可从 SMILES 直接捕捉原子级别电子与空间效应;
能同时预测立体选择性与绝对构型,适用于复杂的双前手性体系;
展现出强外延能力,可迁移至不同类型的不对称反应;
解释性强,有利于催化剂与底物设计。
未来改进方向包括:
扩充底物与催化剂类型以减少数据偏倚;
融入三维信息以捕捉构象效应与弱相互作用;
引入更丰富的反应条件(温度、压力、溶剂参数等);
在新配体与金属体系中进行实验验证;
创建更系统化的跨领域基准测试。
总体而言,ChemAHNet 为不对称催化的预测与设计提供了强有力工具,为数据驱动的催化开发奠定了坚实基础。
整理 | DrugOne团队
参考资料
Cheng, L., Shao, PL., Lv, J. et al. Chemistry-informed deep learning model for predicting stereoselectivity and absolute configuration in asymmetric hydrogenation. Nat Comput Sci (2025).
https://doi.org/10.1038/s43588-025-00920-8

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢