
本文介绍了澳门理工大学刘焕香教授和姚小军教授团队提出的一种激酶-抑制剂活性与选择性预测新方法。由于激酶结构的高度保守以及激酶组学实验成本高昂,高选择性的激酶抑制剂开发面临巨大挑战。因此,准确预测激酶-抑制剂的亲和力与特异性尤为重要。在这项研究中,作者提出了一个注意力一致性引导的对比学习框架--MMCLKin。该方法融合了几何图网络与序列网络,并结合多头注意力机制以及多模态、多尺度对比学习方法实现对激酶-抑制剂活性与选择性精确且可解释的预测。MMCLKin在两个三维激酶-药物数据集上的预测表现优于现有方法,并在十种不同的蛋白质-药物数据集及一个突变敏感数据集上展现出良好的泛化能力,同时也能够有效应用于结构已知或未知的激酶筛选中。注意力分析也进一步表明MMCLKin能够准确识别对激酶-抑制剂结合至关重要的关键残基与分子功能基团。此外,ADP-Glo实验证实,在MMCLKin驱动筛选出的20个候选化合物中,有5个能够抑制致病性LRRK2 G2019S突变体,其中4个表现出纳摩尔级别的抑制活性。总得来讲,MMCLKin为高效且高选择性的激酶抑制剂发现提供了一个有效工具。

研究背景
激酶是调控众多细胞功能的关键蛋白,其功能异常与癌症、自身免疫病等多种重大疾病密切相关,是21世纪最重要的药物靶点之一。然而,由于激酶家族结构高度相似(尤其是ATP结合位点),开发兼具强效性与高选择性的抑制剂面临巨大挑战。许多候选化合物因脱靶效应而止步于临床阶段。尽管激酶组实验分析方法结果精准,但成本极高、通量有限,难以用于大规模筛选。因此,发展高效、准确的预测方法成为加速发现新型激酶抑制剂的关键。
方法
首先,本研究提出了MMCLAC方法,通过层次化对比学习整合异质信息。使用双维度对比机制:
跨模态对比:对齐序列表征与三维图结构的注意力分布,同步捕获序列信息、上下文特征与空间结构,缓解激酶序列高自由度和图稀疏性问题。
跨尺度对比:协调局部原子级与全局结构级注意力,既关注关键药物-口袋相互作用,又保持整体激酶信息感知。
此外,该方法使用节点级对比学习实现注意力一致性,不仅促进同一体系内不同模态、不同尺度特征的对齐,更增强模型区分不同激酶-药物体系细微差异的能力,从而提升对相互作用特异性与选择性的捕捉能力。
基于MMCLAC方法,我们进一步构建了MMCLKin框架(图1),包括:
3D数据集的构建(图1a):基于AlphaFold2预测结构,提取高置信度的激酶结构域与关键结合位点,并通过LigPrep优化小分子构象至最低能量状态,进而构建三维3DKDavis与3DKKIBA数据集,降低数据噪声。
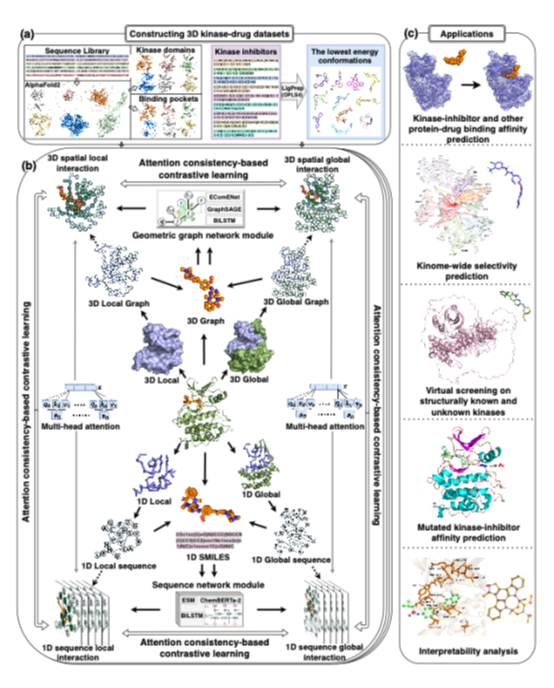
图 1:MMCLKin 的整体框架示意图。
特征提取模块:
几何图网络:兼顾局部与全局完整性,全面捕捉激酶空间结构与小分子三维构象(图2a)。
序列网络:融合蛋白语言模型与化学语言模型,提取激酶序列进化信息与小分子精细化学特征(图2b)。
整合与预测:
多头注意力机制自主学习不同作用范围的激酶-抑制剂依赖关系,量化各要素贡献(图2c)。
MMCLAC对齐节点级注意力分布,确保空间、序列、局部、全局特征有效整合(图2e)。
预测模块融合跨模态、跨尺度相互作用信息生成最终结果(图2d)。
应用:
MMCLKin在多种应用场景中均表现出色(图1c),包括激酶-抑制剂及其他蛋白-药物亲和力预测、激酶抑制剂选择性分析、已知结构/未知结构/突变激酶的虚拟筛选,以及可解释性分析。
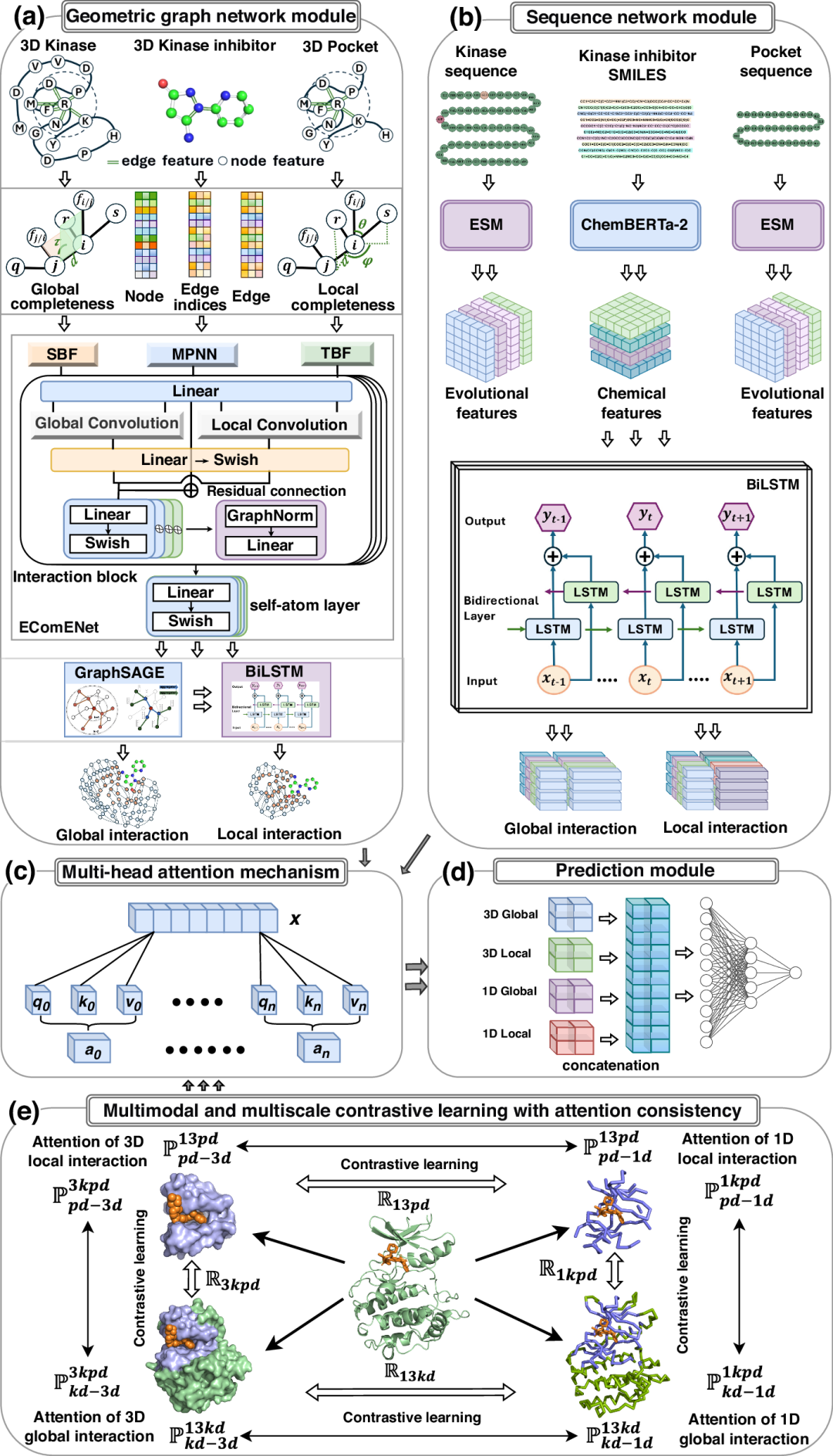
图 2:MMCLKin 框架
结果与讨论
MMCLKin 在激酶–药物亲和力预测中表现出稳健的性能
ConPLex 在 3DKKIBA 和 3DKDavis 上的五项指标均优于原始数据集(KIBA 和Davis),尤其在 CI、PCC 和 Spearman 系数上提升显著,说明构建的数据集能够更有效地帮助模型捕获关键信息(图3a和3b)。
我们随后在构建的两个数据集上,基于三种划分策略评估了 MMCLKin 的预测性能。比较对象涵盖三类基线模型:序列模态(TransformerCPI、FusionDTA、PSICHIC)、二维分子图(GraphDTA、DrugBAN)以及三维结构表征(KDBNet)。在 3DKDavis 数据集上,MMCLKin 在激酶冷启动和激酶–药物冷启动两种划分下的四项指标均优于所有基线模型,且预测方差更低(图3c)。在药物冷启动设置中,MMCLKin 在 MAE、MSE 和 CI 上亦取得最佳表现。在 LSKIBA 数据集(3DKKIBA中的低相似度子集,仅包含 Tanimoto 相似度低于 0.4 的化合物对,图3d)上,MMCLKin 在三种划分策略下的 MAE 和 MSE 均持续领先。此外,五折交叉验证也进一步证明MMCLKin 在激酶–药物亲和力预测任务中保持了高度竞争力。
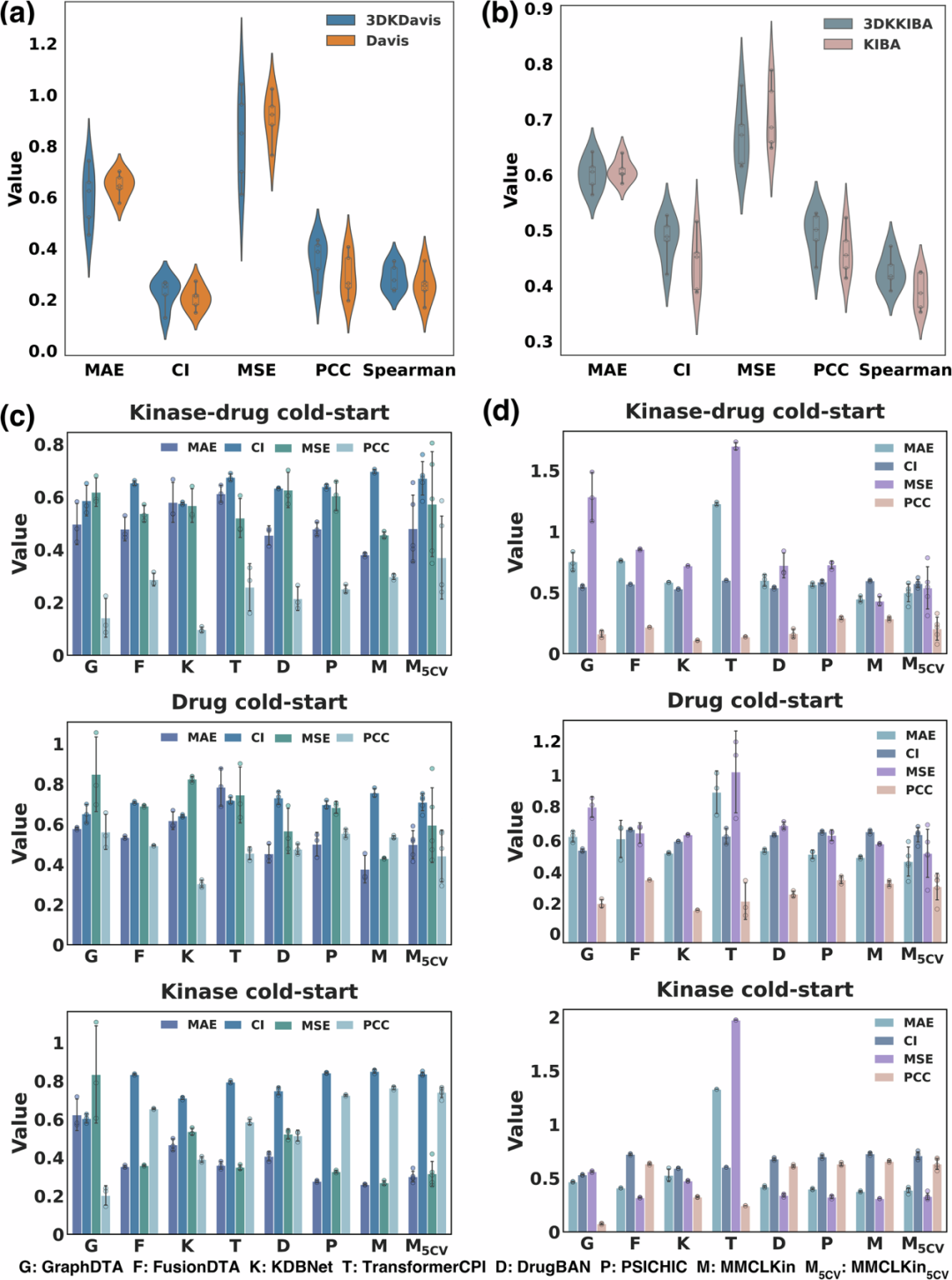
图 3:MMCLKin 在两个构建的 3D 数据集上针对药物冷启动、激酶冷启动及激酶-药物冷启动划分策略的亲和力预测性能表现。
MMCLKin 在人类激酶组中的抑制剂选择性预测表现优异
图4 展示了 MMCLKin、FusionDTA、TransformerCPI 和 KDBNet 在两类数据集上的选择性指标预测值与真实值之间的 Pearson 相关性。在 3DKDavis 数据集上,MMCLKin 在standard score, Gini coefficient和selectivity entropy上的相关性分别达到 0.898、0.537 和 0.651,显著优于所有基线模型,表明其预测选择性更加接近真实情况。在低相似度的 LSKIBA 数据集上,MMCLKin 在全部指标上均取得最高或相当的相关性,进一步体现出其稳健性与强泛化能力。
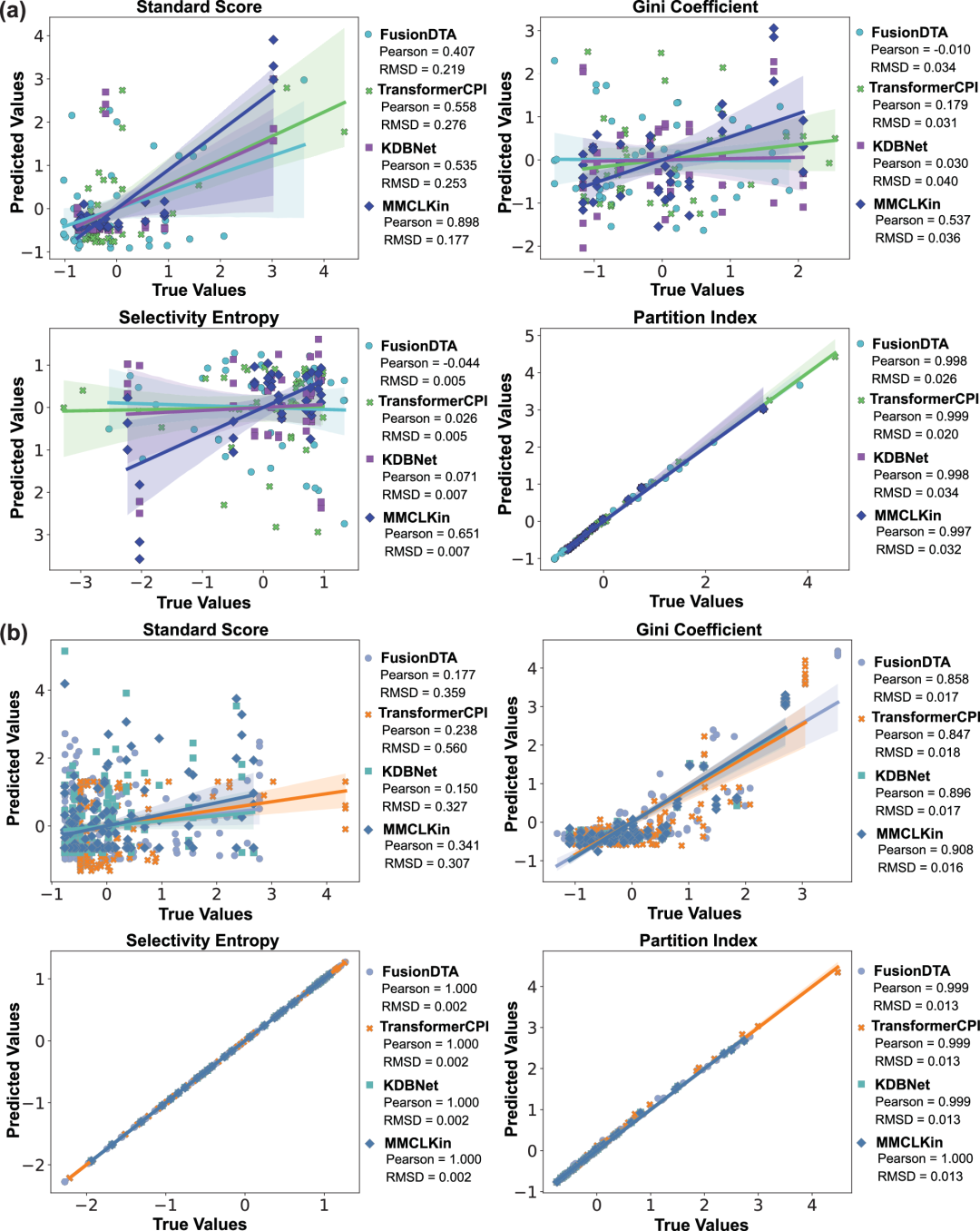
图 4:MMCLKin 在全人类激酶组范围内的激酶抑制剂选择性预测性能表现。
MMCLKin 在多样化的蛋白结构上展现出良好的泛化能力
表1对比了 MMCLKin 与已有模型在 CASF-2016 测试集上的表现。结果显示,MMCLKin 在三项指标上均优于所有无复合物(complex-free)模型;与基于复合物的模型相比,MMCLKin 亦表现出高度竞争力,其 RMSE 和 MAE 与最佳模型 IGN 接近。
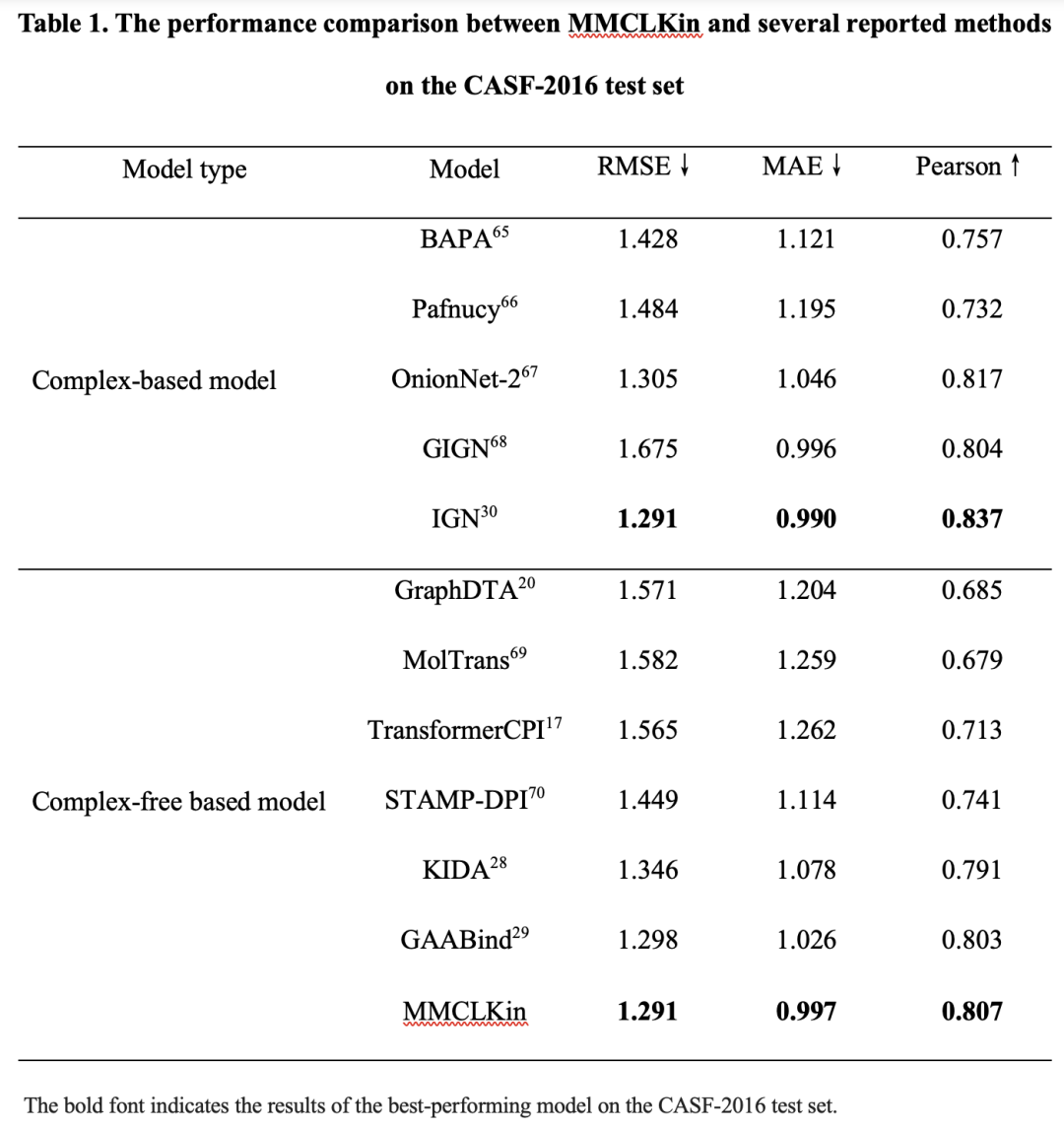
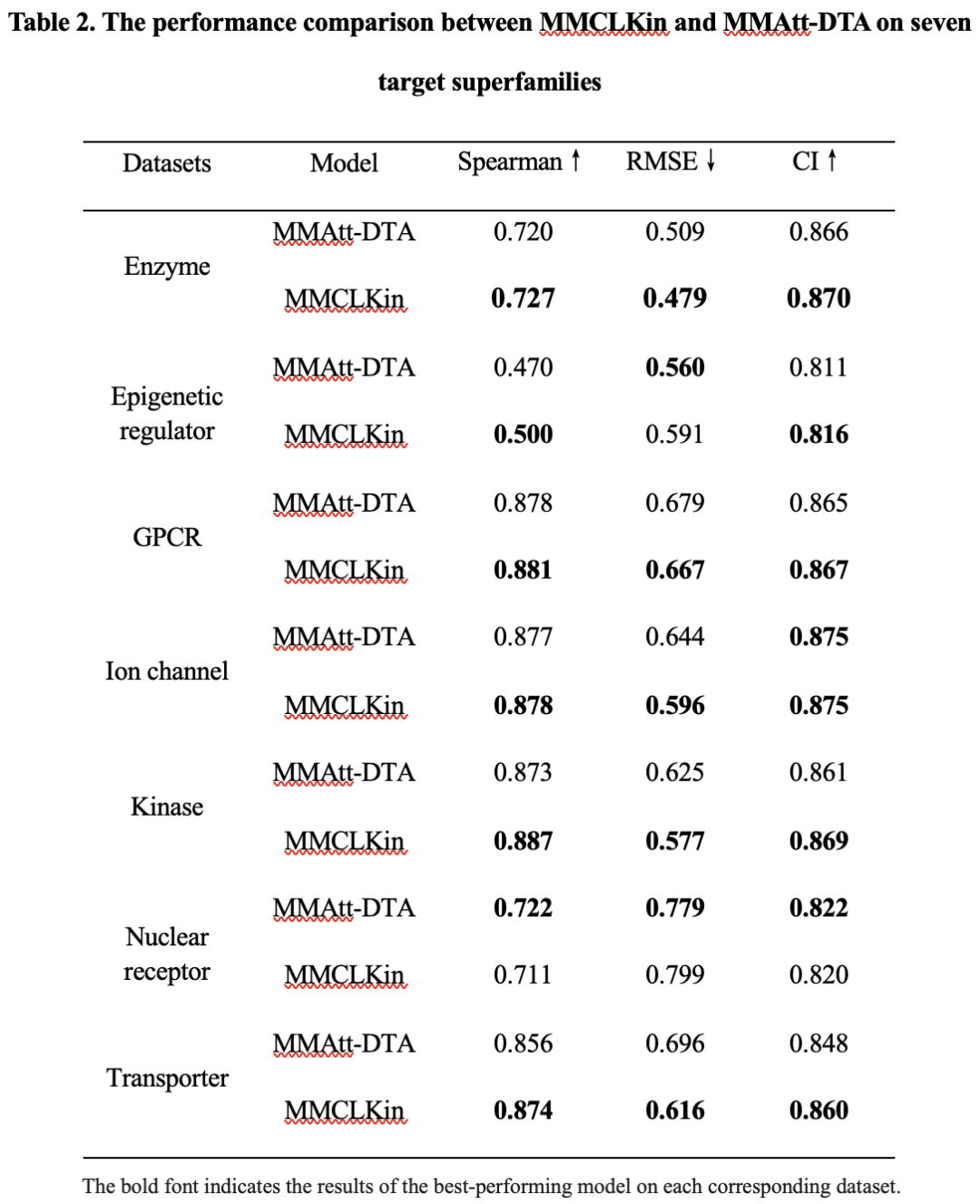
表 2 总结了 MMCLKin 与 MMAtt-DTA 在七类靶标超家族上的性能对比。MMCLKin 在其中六个数据集上优于 MMAtt-DTA,并在 Enzyme、GPCR、Ion channel、Kinase 和 Transporter 等数据集上,在 RMSE、CI 和 Spearman 相关系数方面均取得最佳表现。
MMCLKin 在具有实验解析 3D 结构的两个激酶靶点上展现出强劲的虚拟筛选性能与良好的可解释性
与 Glide SP 分子对接方法相比,MMCLKin在识别LRRK2和HPK1的活性抑制剂方面均表现出显著优势。对于LRRK2(图5b),在排名前1%的化合物中,Glide SP 的召回率为 36.36%,而MMCLKin达到45.45%;在前5%与10%阈值下,MMCLKin 的召回率分别为72.73%和81.82%,显著高于Glide的45.45%。在HPK1(图5c)上,MMCLKin在前1%、2%和10%的召回率分别达到42.86%、57.14%和64.29%,同样超过Glide的28.57%、35.71%和42.86%。
为评估模型可解释性,我们将注意力权重映射回激酶靶点的残基及其代表性抑制剂的重原子上,并聚焦注意力值最高的15个残基。对于 LRRK2(图5d),Val1946、Glu1948、Ala1950、Kys1952 和 Ser1954位于关键的铰链区域,其中Glu1948与Ala1950是多数抑制剂形成氢键的核心位点。Glu1902、Ala1904、Val1905分布于N端小叶β折叠区,Leu2001、Leu2002、Phe2003、Ile2015位于C端大叶β折叠区。对于HPK1(图 5e),Glu92 位于铰链区域,Arg22、Leu23、Gly25、Val31、Val43、Ala44、Leu45、Ile89、Cys90 位于小叶 β 折叠区,Leu144、Asn142、Arg152、Leu153 位于大叶 β 折叠区。上述区域均被广泛证实为决定激酶–抑制剂特异性与结合稳定性的关键结构。
此外,MMCLKin 对抑制剂分子中的极性官能团(如羟基、氨基、亚胺基、三级胺、吡咯环和酰胺基)也给予显著关注。这些基团通常与激酶结合口袋内残基形成强极性相互作用,从而增强亲和力与选择性。
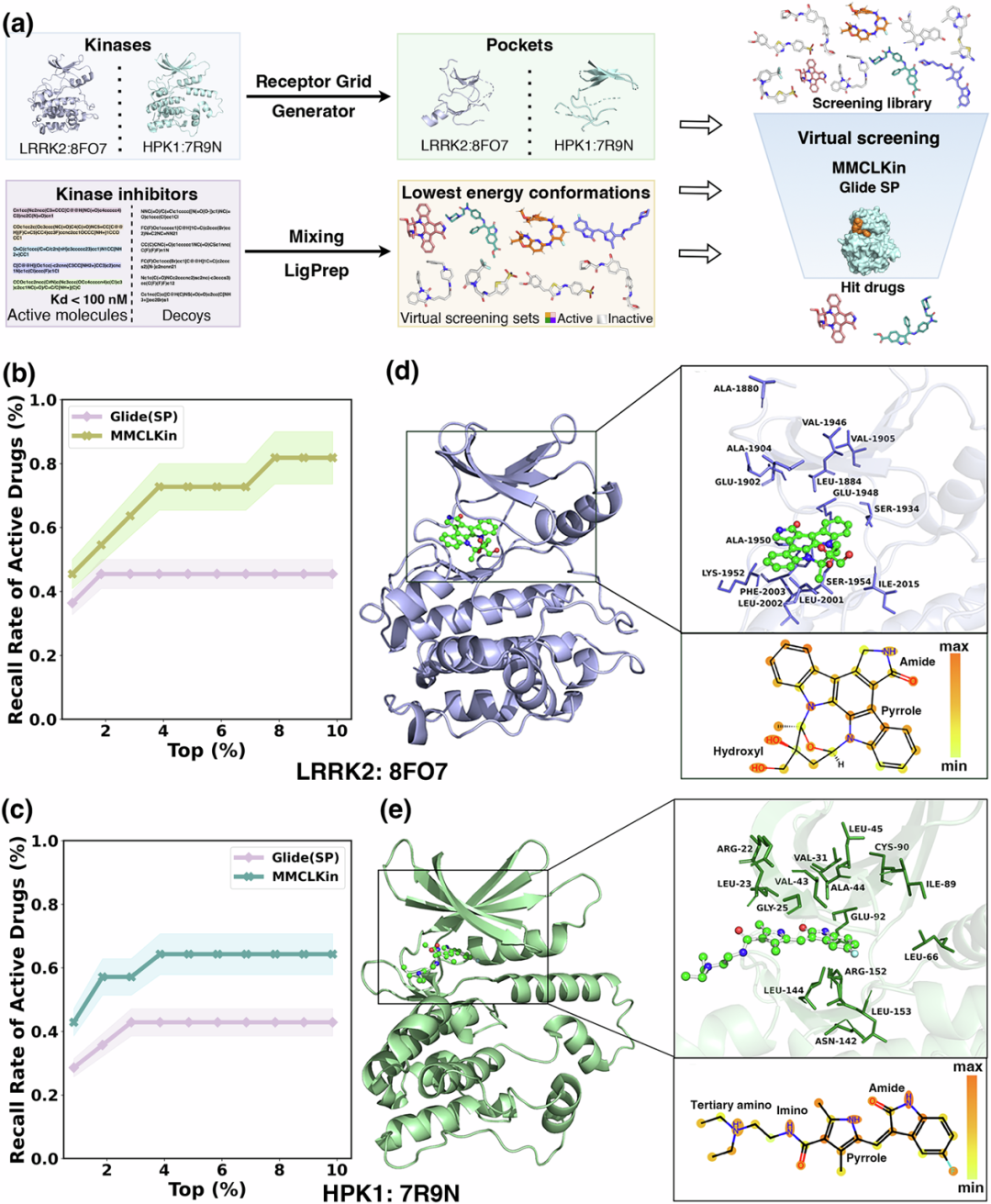
图 5:MMCLKin 在具有已知实验结构的两类激酶(LRRK2 与 HPK1)上的虚拟筛选性能与可解释性分析。
MMCLKin 在缺乏实验 3D 结构的两种激酶靶点上仍保持出色的虚拟筛选性能和可解释性
即使在缺乏实验解析的激酶 3D 结构情况下,MMCLKin 仍保持出色且更具竞争力的虚拟筛选性能。对于 NUAK2(图6b),MMCLKin 在排名前 2%、5% 和 10% 的化合物中召回率分别达到 41.67%、50% 和 66.67%,明显高于 Glide SP 的 25%、33.33% 和 41.67%;对于 CRK12(图6c),在排名前 5% 和 10% 的召回率分别达到 53.85% 和 84.62%,远超 Glide SP 的 15.38% 和 23.08%。
在可解释性方面,MMCLKin 对预测的激酶结构同样表现稳健。NUAK2 的前 15 个关键残基中,铰链区域的 Glu130、Tyr131、Ala132、Arg134 和 Asp136 被优先关注(图6d);CRK12 中铰链区域的 Pro431、Tyr432 和 Ala433(图6e)也同样被关注。其他关键残基主要分布于 N、C 叶 β 折叠结构,进一步显示 MMCLKin 在预测结构上的泛化能力。此外,MMCLKin 依然对抑制剂的极性官能团(氨基、醚、酮羰基、吡咯和酰胺)保持高度关注,这些官能团易与结合口袋形成氢键或静电相互作用,从而增强激酶–药物相互作用的特异性与结合强度。
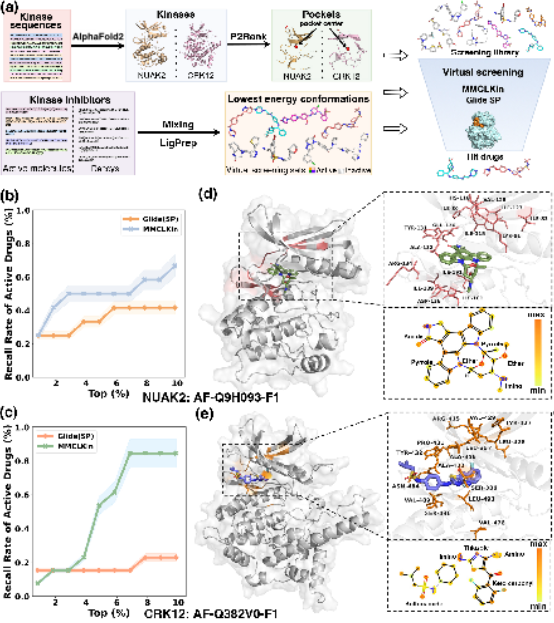
图 6:MMCLKin 在无实验解析结构的 NUAK2 与 CRK12 上的虚拟筛选性能与可解释性分析。
MMCLKin驱动的LRRK2 G2019S抑制剂发现与活性验证
为评估 MMCLKin 在突变激酶靶向药物发现中的实际应用,我们将 MMCLKin 虚拟筛选与 ADP-Glo 实验验证相结合。结果显示,在筛选获得的 20 个候选化合物中,有 5 个化合物(LY2025-01 至 LY2025-05)在 10 微摩浓度下显示出超过 50% 的抑制率,其中 4 个达到纳摩尔级抑制活性。
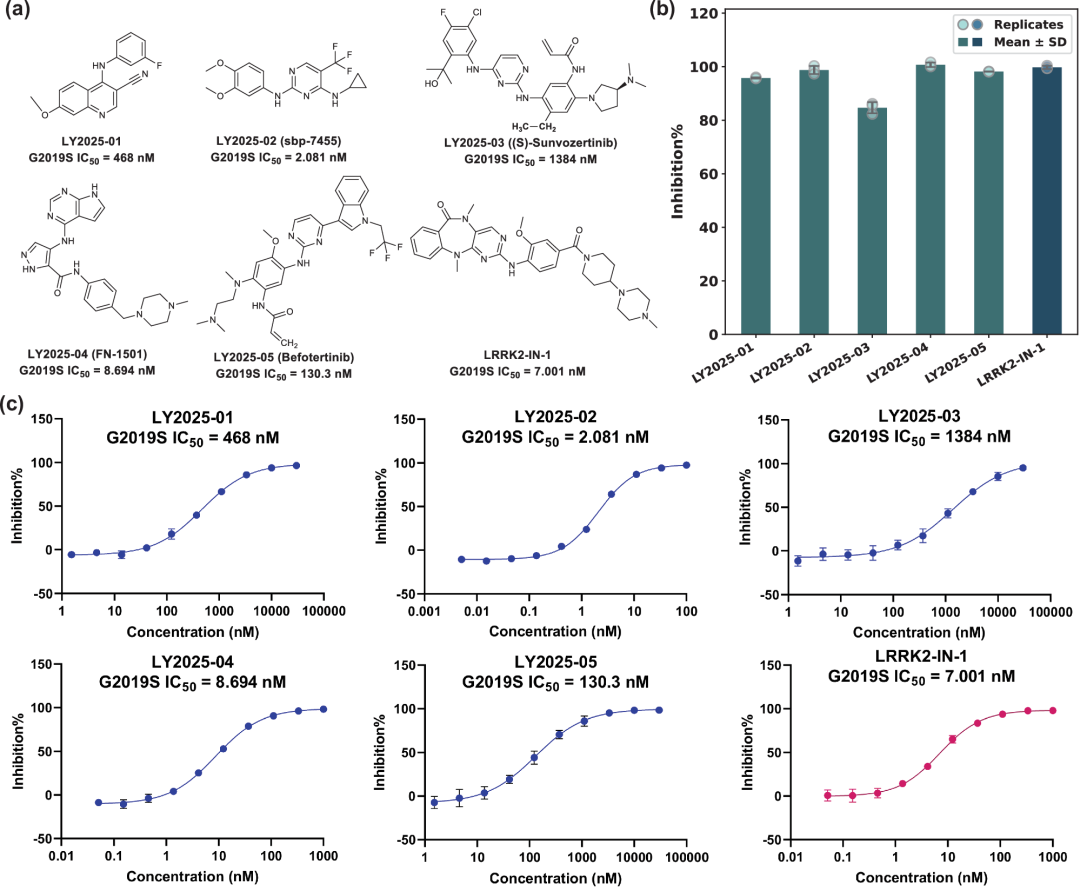
图7:MMCLKin 鉴定的五种化合物及阳性对照的化学结构,以及其在 10 μM 浓度下的抑制率和 IC₅₀ 数值
结论
本研究提出了MMCLKin预测框架,通过几何图网络与基于大语言模型的序列网络提取特征,并结合多头注意力机制和基于注意力一致性的多模态多尺度对比学习方法对各类相互作用特征进行有效整合。实验结果表明,MMCLKin在两个三维激酶-药物数据集上的预测表现优于现有方法,在十个蛋白结构数据集及一个突变感知数据集中展现出优异的泛化能力。同时,该框架对结构已知、未知及突变激酶靶点均具有良好筛选能力,且能自主识别关键结合残基与功能基团,体现了模型的可解释性。此外,ADP-Glo实验结果显示,在MMCLKin驱动下筛选的20个候选化合物中,5个对LRRK2 G2019S突变体具有抑制作用,其中4个化合物的活性达到纳摩尔级别。综上,MMCLKin为高选择性、高亲和力激酶抑制剂的发现提供了有效的计算工具,并展现出拓展应用于其他蛋白家族的潜力。然而,尽管多尺度多模态特征的整合提升了模型性能,但也带来了较高的计算需求。未来研究需进一步优化特征提取与融合策略,在保证预测精度的同时提升计算效率。
参考资料
Tian, Y., Lu, R., Gong, X. et al. Enhancing kinase-inhibitor activity and selectivity prediction through contrastive learning. Nat Commun 16, 10860 (2025).
https://doi.org/10.1038/s41467-025-65869-8
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢