
靶向 RNA 的小分子药物被视为调控细胞通路与治疗复杂疾病的重要新策略,为非蛋白靶向药物开发提供广阔空间。然而,现有方法在小分子配体的发现能力与适用性上仍存在显著限制。诸如AlphaFold3等通用结构模型虽展现出预测RNA-小分子复合物结构的潜力,但对于结合强度预测以及指导药物筛选仍存在鸿沟。
近日,上海交通大学溥渊未来技术学院/人工智能学院郑双佳课题组联合广东工业大学陈修财教授团队, 南京理工大学於东军教授团队以及来自哈佛大学,麦吉尔大学,中山大学等高校的研究人员共同提出一种面向RNA-小分子结合预测的通用型几何深度学习框架GerNA-Bind。研究通过将RNA多模态信息与不确定性评估引入结构大模型体系,实现了对RNA-配体相互作用更加稳定、可信、可解释的预测。在一项湿实验验证中,研究团队利用GerNA-Bind针对肿瘤相关的RNA靶点MALAT1进行构象特异的高通量虚拟筛选,鉴定出了18个与其结合的分子;其中结合效果最好的化合物经过验证能够特异性靶向MALAT1三螺旋结构,并在细胞实验中展示出抑制癌细胞迁移的能力,证明了该模型的实战能力。这一工作旨在突破RNA靶向药物研发长期存在的瓶颈,让结构驱动的AI方法真正支撑药物筛选与机制分析。

模型架构
GerNA-Bind以RNA与配体结构为输入,RNA可来自实验解析或通过RNA折叠方法预测,配体亦包含对应的3D构象。GerNA-Bind具备高准确性、稳健性与可解释性,可用于RNA-配体结合特异性预测、靶向筛选、结合位点定位及作用机制分析。
GerNA-Bind采用多状态表示同时整合RNA与配体的1D序列、2D图结构与3D构象,并以序列编码器、图神经网络和等变图Transformer进行分层建模。在此基础上,模型通过几何约束模块对RNA与配体在2D拓扑与3D空间中的相互作用进行物理约束,最终输出结合亲和力用于估计特异性。同时引入基于证据的 Dirichlet 分布估计概率与不确定性,为筛选决策提供可靠性信息。
此外,GerNA-Bind 通过几何约束模块进一步优化碱基—原子成对接触矩阵,使得相互作用预测更加精细且具可解释性,为结合特异性判断提供直接结构依据。
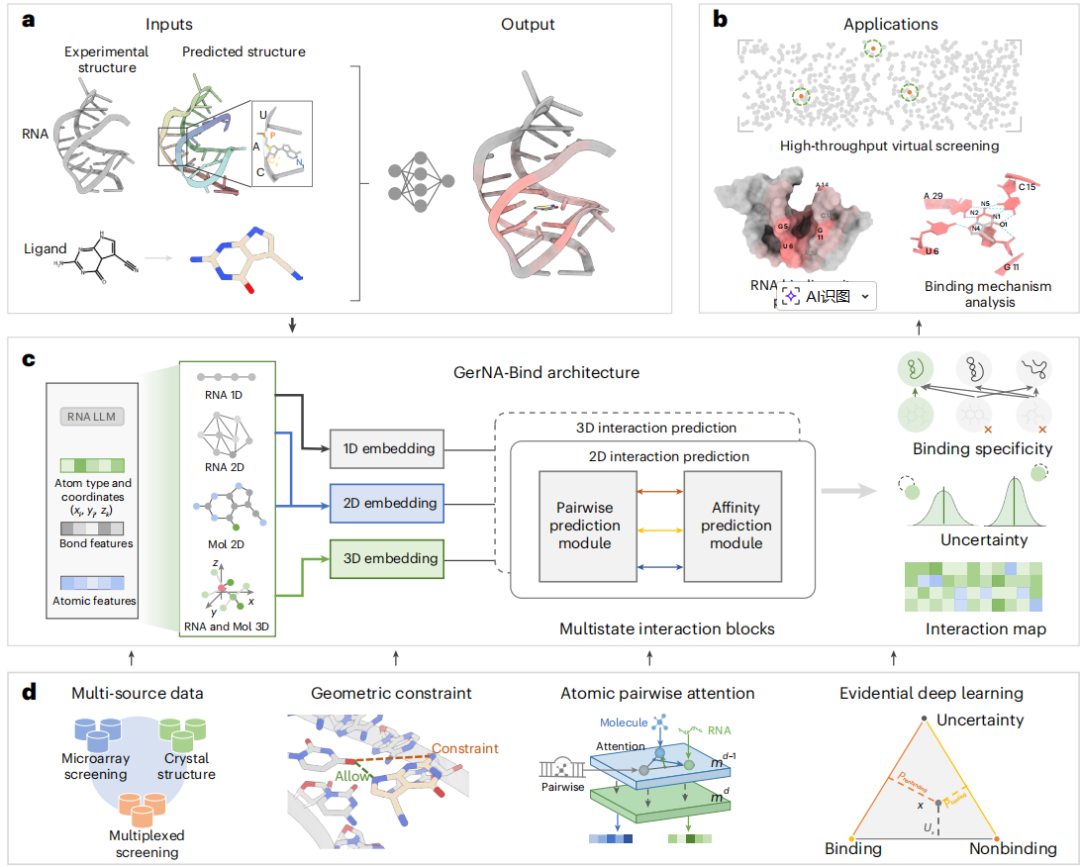
图1. 模型架构图。GerNA-Bind是一种多模态的几何基座模型用于RNA-小分子特异性结合预测并可应用于靶向RNA的高通量虚拟筛选。
RNA–配体结合特异性预测的准确性
研究者在两个公开数据集(Robin 与 Biosensor)以及四种拆分策略上评估 GerNA-Bind。总体而言,GerNA-Bind 显著优于现有方法:在 Robin 数据集的同源/指纹拆分中,比次优模型 GraphDTA 提高 6.7% AUROC,比 RSAPred 提高 12.4%;在 Biosensor 数据集中则分别提高 9.1% 与 12.6%。多模态输入带来的增益明显:相较于单模态模型,GerNA-Bind 在 Biosensor 上 AUROC 提升 14.4%、在 Robin 上提升 8.6%;即便在最苛刻的拆分下仍保持显著提升(分别为 9.1% 与 5.5%),表明模型在低同源/外域场景中的鲁棒性和泛化能力。
GerNA-Bind 内建基于证据的不确定性估计,可为每个预测分配置信度。以 Robin 数据集为例,将预测按不确定性排序并计算累积 AUROC,GerNA-Bind 显示出与性能高度相关的置信评估;在随机拆分实验中,不确定性与 AUROC 的 Spearman 相关系数达 0.96,明显优于集成学习(0.79)和 dropout(0.82)。该特性使得我们能以置信度为筛选标准,在虚拟筛选中优先验证高可信候选,从而提高实验命中率与资源利用效率。
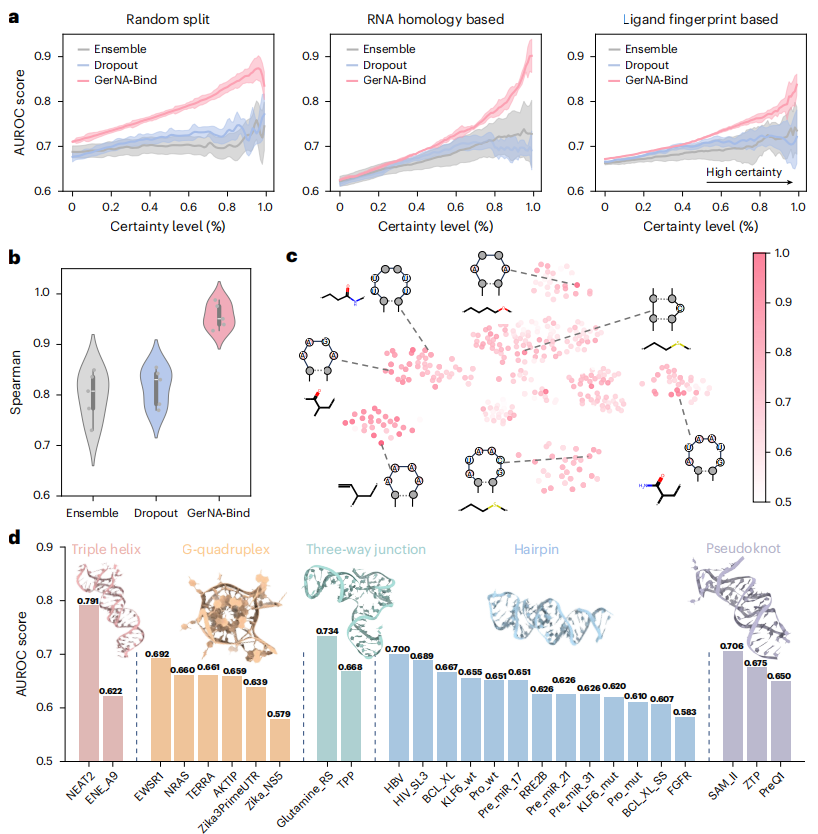
图2. 模型不确定性估计能力以及其在不同类型的RNA-小分子互作预测能力。
结合位点预测与模型可解释性
GerNA-Bind 通过几何约束模块直接预测碱基—原子成对接触矩阵,实现结合位点的定位。经 Hariboss 结构集微调后,结合位点预测的AUROC从无监督时的 0.557 提升至 0.808,并在与 RNASite、Chai-1、AlphaFold3 的比较中,在 Precision、F1、AUPRC、MCC 等指标上分别实现约 20.8%(Precision)/13%(F1)/10.6%(AUPRC)/15.6%(MCC) 的优势。在不同 RNA 结构类别的独立测试中,模型在多数类别中取得最高 AUROC,且预测的结合位点与真实结合位置一致,证明其在机制解析上的实用性。
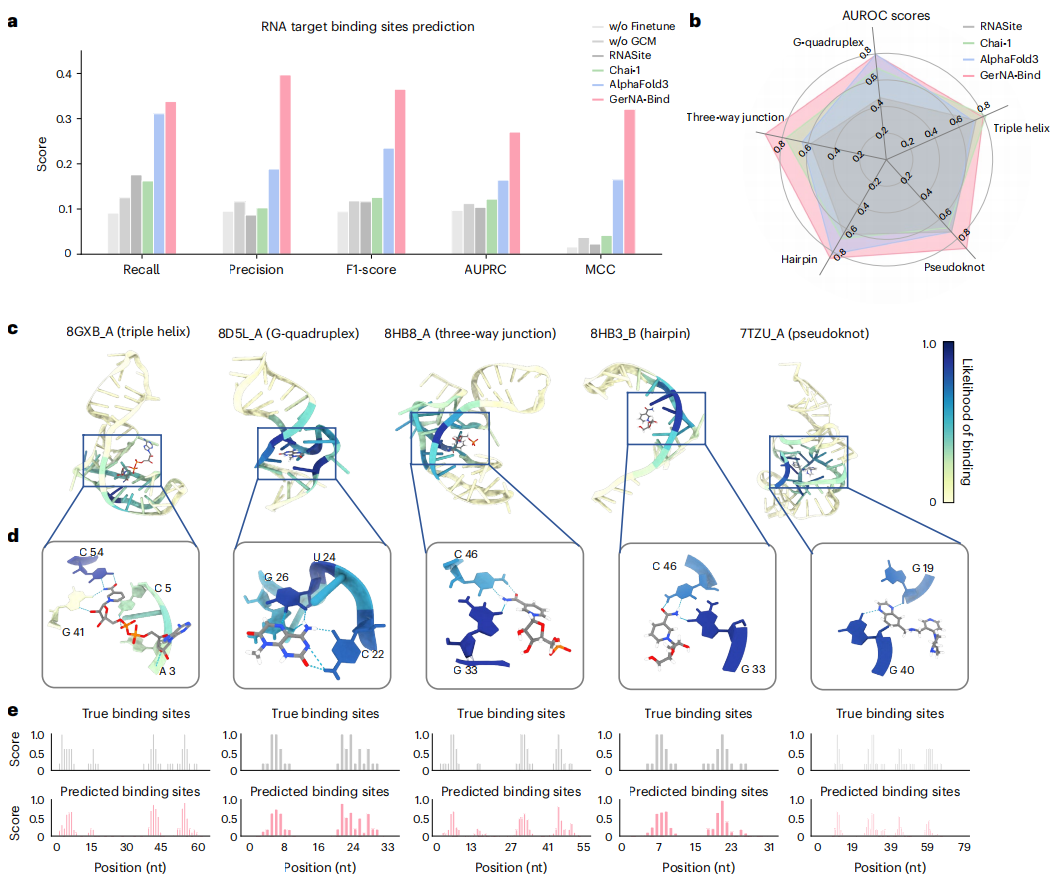
图3. 模型相比起AlphaFold3等模型对于结合位点的预测更加准确。
对 MALAT1 三链结构的大规模虚拟筛选与实验验证
研究者们基于 Topscience 库构建了21,659个RNA-小分子化合物库,并用 GerNA-Bind 进行打分与不确定性筛选,最终挑选出 28 个优先候选。经 thiazole orange 位移实验验证,28个候选中18个(64%) 显示>50%位移,证明其发生结合;其中 Mol14、Mol10、Mol6 的位移率分别为 80.9% / 69.8% / 69.3%(参考阳性化合物为68.4%)。Mol14与已知的跟MALAT1结合的化合物相似度低(Tanimoto=0.17),表现出化学骨架的新颖性;竞争性位移与突变验证表明 Mol14 选择性识别 MALAT1 的三链位点(DC₅₀ = 58 nM),并在 A549 细胞中降低 MALAT1 表达且抑制细胞迁移。该案例展示了 GerNA-Bind 在湿实验验证闭环中的实际可用性与转化潜力。
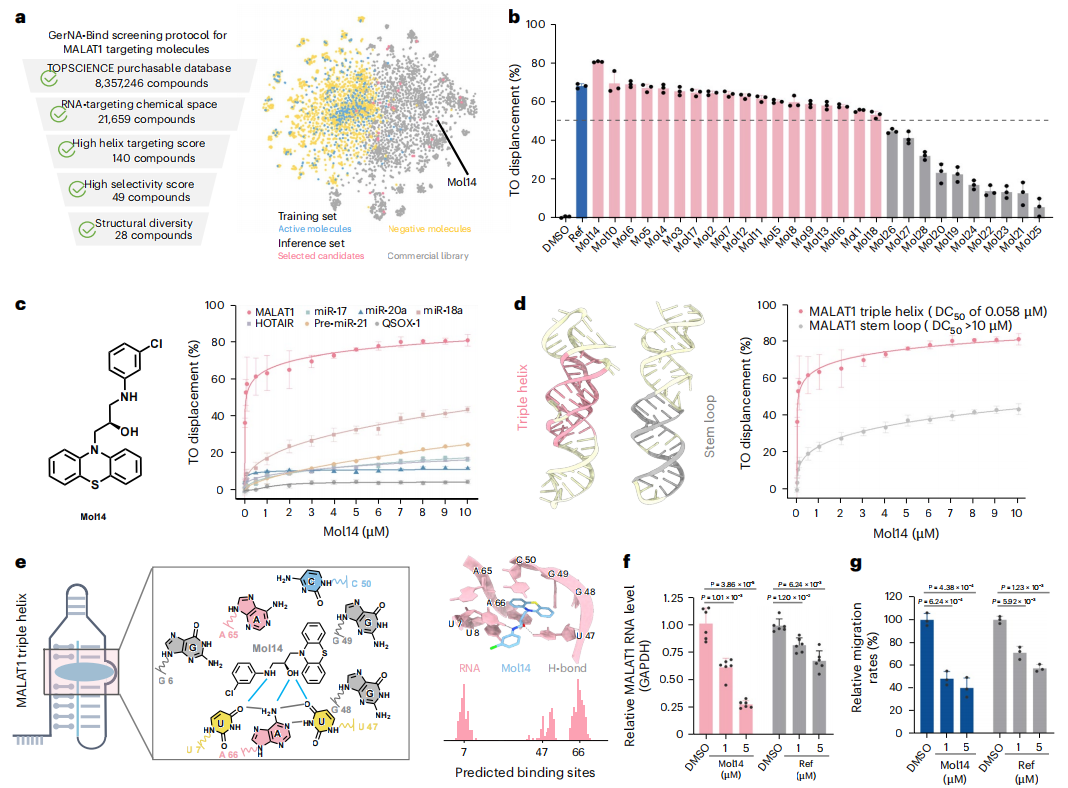
图4. 针对多构象RNA癌症靶点MALAT1进行构象特异的虚拟筛选并鉴定出18个结合分子,其中4号分子展现出三螺旋特异靶向性并在细胞实验上验证了其抑制细胞迁移活性。
讨论
研究人员提出了 GerNA-Bind,一个用于解析RNA-小分子相互作用的几何深度学习框架,旨在提升 RNA 靶向虚拟筛选的效率与可靠性。相比依赖高通量筛选或缺乏可解释性的深度学习方法,GerNA-Bind通过显式建模RNA-配体接触模式,实现更高性能、更强泛化性与更清晰的机制洞察。
其核心优势包括:
多模态整合:融合 RNA 与小分子的 1D、2D、3D 多模态表征;
显式相互作用建模:以逐碱基、逐原子接触矩阵提供可解释机制;
不确定性估计:模型同步提供不确定性估计,用于提升准确性与预测置信度;
计算与实验双重验证:在公开数据集上达成领先性能,并实验证实可筛选出针对MALAT1的多样化hits和先导分子。
当前限制主要在于模型专注RNA-小分子体系,尚未覆盖RNA-蛋白或复杂多聚体,也暂不支持修饰碱基;同时,高质量 RNA-配体数据仍相对不足。未来可通过整合更多结构工具、引入迁移学习与结构集合来扩展模型的适用性与稳健性。
总体而言,GerNA-Bind 以多模态几何结构为核心,兼具预测性能、可解释性与泛化能力,为 RNA 靶向药物发现提供了坚实的计算基础,并为生成式药物设计和更广泛的分子互作建模打开新的发展方向。
参考资料
Xia, Y., Li, J., Chu, YT. et al. Deciphering RNA–ligand binding specificity with GerNA-Bind. Nat Mach Intell (2025).
https://doi.org/10.1038/s42256-025-01154-z
代码
https://github.com/GENTEL-lab/GerNA-Bind
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢