
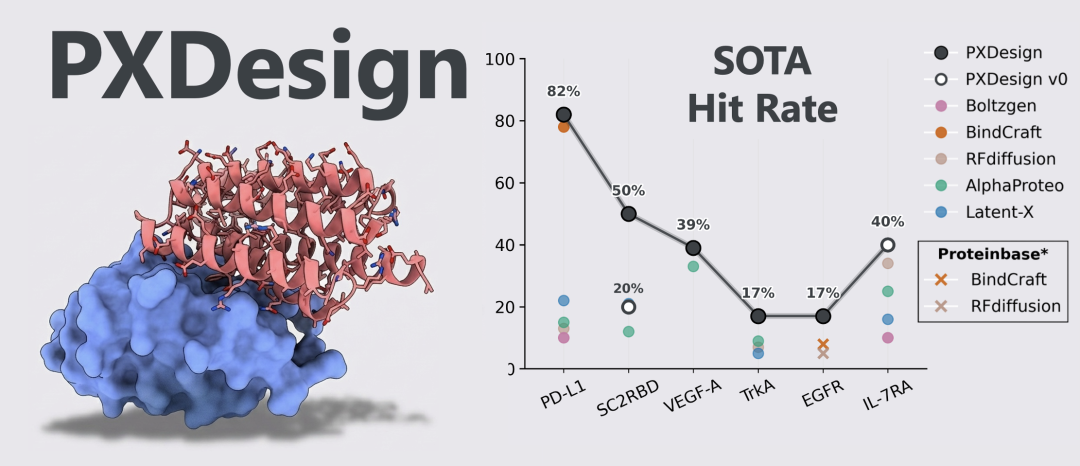
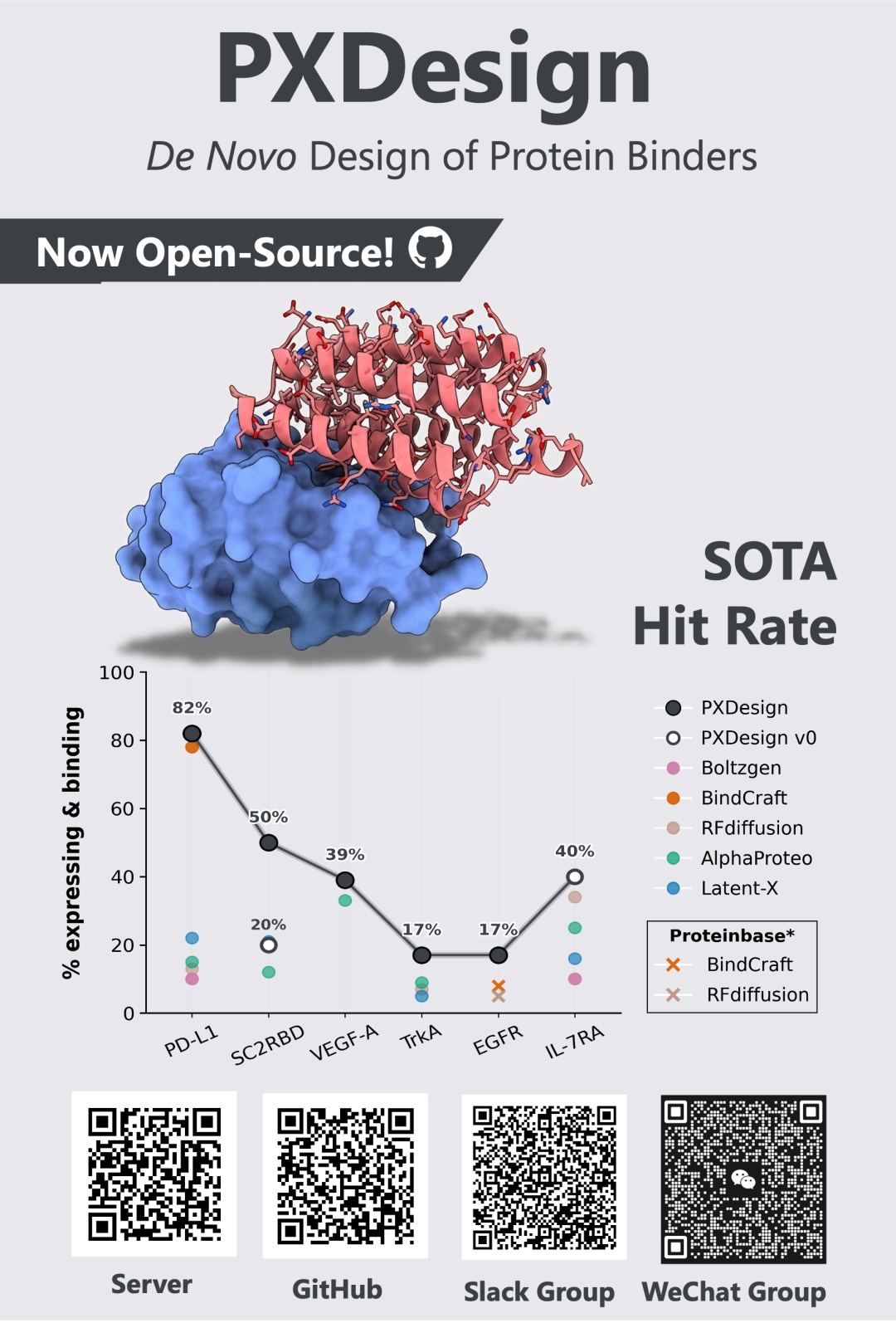
PXDesign的用途
PXDesign 是一套面向 de novo protein binder 的自动化设计pipeline,核心目标是提高“从计算设计到湿实验命中”的成功率。在针对 7 个真实蛋白靶点开展的系统实验验证中,PXDesign 在其中 6 个靶点上取得了 17%–82% 的纳摩尔级 binder 实验成功率。
对于第一次接触 PXDesign 的湿实验背景的科学家,也可以优先使用 PXDesign Web Server快速体验。
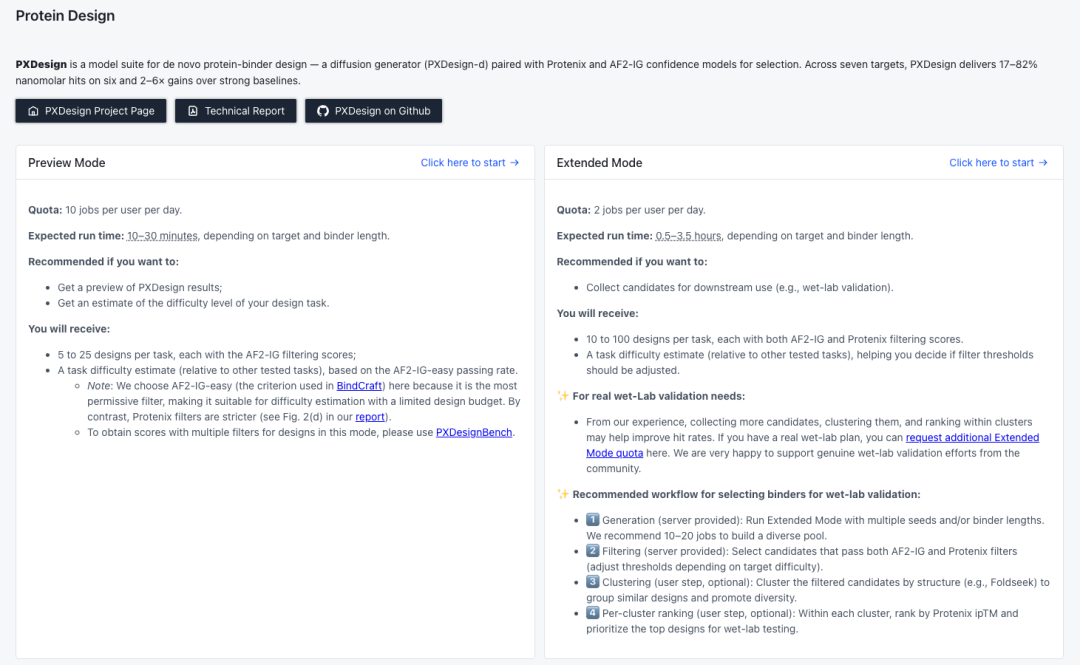
因为 PXDesign Web Server 无需 GPU和安装配置,运行流程与论文完全一致,且已在大量真实科研项目中得到验证,非常适合用于高效获取高置信度 Binder 候选,并避免将时间浪费在复杂环境搭建上。
PXDesign 本地版安装方法
首先,将 PXDesign 仓库克隆到本地:
git clone https://github.com/bytedance/PXDesign.git
cd PXDesignPXDesign 提供了一个一键安装脚本 install.sh,自动完成依赖配置:
bash -x install.sh --env pxdesign --pkg_manager conda --cuda-version 12.1
安装完成后,激活环境:
conda activate pxdesign运行以下命令检查 PXDesign 是否可用:
pxdesign pipeline --help如果能看到参数说明,说明安装成功 🎉
首次运行前需要下载 PXDesign 需要的外部工具的模型权重,需要手动下载一次。
bash download_tool_weights.shPXDesign 本地版用法
使用PXDesign设计binder需要.cif 或 .pdb 格式的靶点结构,同时需要明确作为结合目标的多肽链(chains) 和 序列范围(crop) 和 希望binder结合的区域(hotspots) ,同时为了Protenix能正常预测靶点蛋白结构,extend mode还需要靶点蛋白的MSA,以及需要指定binder长度。
PXDesign 所有设计任务,都由一个 YAML 文件定义,例如:
target:
file: "./examples/5o45.cif"
chains:
A:
crop: ["1-116"]
hotspots: [40, 99, 107]
msa: "./examples/msa/PDL1/0"
binder_length: 80在正式跑任务前,可以先通过pxdesign check-input 检查 YAML 是否写对,以发现YAML 语法错误和字段缺失:
pxdesign check-input --yaml your_task.yaml进一步的,利用pxdesign parse-target可视化检查 crop / hotspot 是否对齐 (强烈推荐)
pxdesign parse-target --yaml your_task.yaml -o debug_dir你会得到*_parsed_target.cif 和 *_parsed_target.pml 文件,下载 debug_dir 目录到本地,用PyMOL打开 .pml 文件,可以看到:
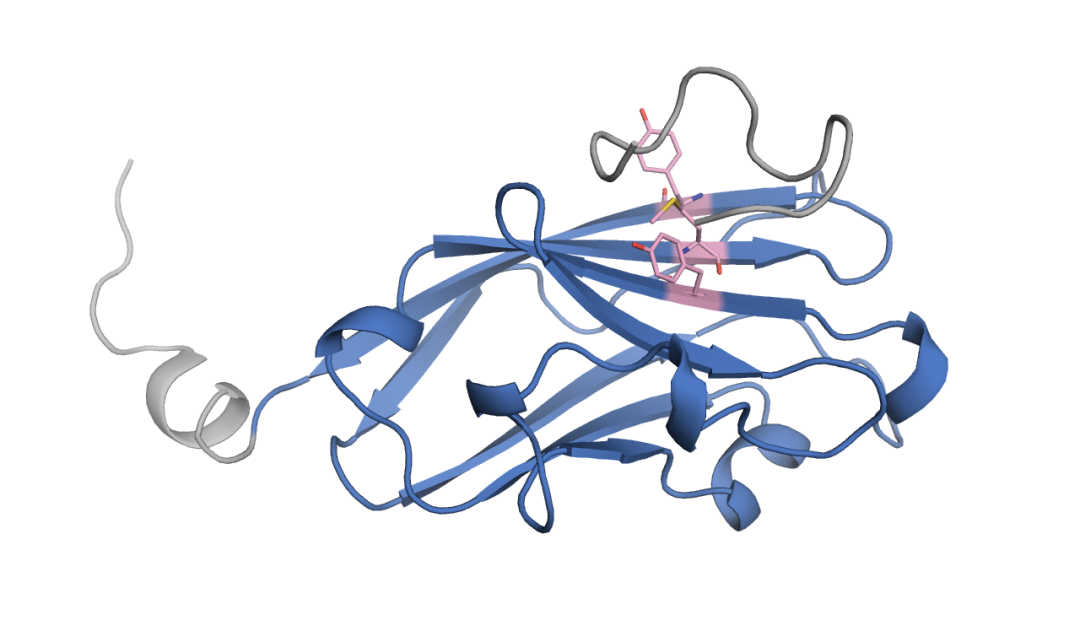
以上信息都确认无误后,可以启动一次extended mode:
pxdesign pipeline --preset extended \
-i <YAML_FILE> \
-o <out_dir> \
--N_sample 500 \
--dtype bf16 \
--use_fast_ln True \
--use_deepspeed_evo_attention True
PXDesign 运行完成后,会在输出目录中生成多个文件和子文件夹。其中:summary.csv 是 所有设计结果的总表,包含了每个 Binder 的核心评估指标和过滤结果。表中重点关注AF2-IG-success,AF2-IG-easy-success,Protenix-success,Protenix-basic-success这些字段。这些列的 True / False ,表示了该设计是否通过对应模型的质量过滤。通常三个True就有实验可行性了。
输出目录中通常会有一张 server_xx_mode.png,用于判断当前设计任务的整体难度。如果任务偏难,考虑增加采样数量、调整 hotspot 或 binder 长度、放宽过滤标准(如使用 Protenix-basic)。 如果任务偏容易,可以严格依赖高置信度过滤结果。
此外,有结构生物学经验的用户可以看通过过滤的结构文件夹,在输出目录中类似:passing-AF2-IG-easy/,passing-Protenix-basic/,这些文件夹中存放的是,已经通过对应过滤标准的 Binder–Target 复合物结构,每个文件通常是一个 .cif 结构,可直接用于可视化或后续分析。
祝愿大家都能获得心仪的binder!
点击阅读原文,访问项目主页,探索更多细节。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢