
 作者:Arash Sadri(原载《J. Med. Chem.》2023年)
作者:Arash Sadri(原载《J. Med. Chem.》2023年)
摘要:基于靶点的药物发现是当前药物研究的主导范式;然而,对其实际效率的全面评估尚属缺乏。本文通过对可追溯至150年前的约32000篇文献和专利进行人工系统性回顾,证明了其明显低效。对所有批准药物起源的分析表明,尽管基于靶点的药物发现已主导数十年,但仅有9.4% 的小分子药物是通过基于靶点的筛选方法发现的。而且,即便是这极小部分药物的治疗效果,也不能完全归因或简化归功于其预设靶点,因为其疗效还依赖于表型观察中无意纳入的脱靶机制。数据表明,还原论的基于靶点的药物发现方法,可能是导致药物研发生产力危机的一个原因。提升效率之道似乎是在选择和优化分子时,优先采用更接近所寻求治疗效应的、更高层级的表型观察(例如利用人工智能和机器学习等工具)。
• 这是对基于靶点的药物发现实际效率的首次系统性全面评估。
• 仅9.4% 的已批准小分子药物通过此方法发现。
• 即使是这些所谓的基于靶点的药物,其治疗效果也依赖于多种脱靶机制。
• 迄今为止,还原论的基于靶点的药物发现效率低下,并可能是生产力危机的一个原因。
• 基于观察性和理论性证据,优先考虑更高层级观察的方法可能更高效。
药物发现处于一个多元科学领域与行业创新发展的交汇点,其终极目标是改善健康。然而,发现新药的效率之低,恐怕会使其他领域的研究人员和从业者感到震惊。获得批准的候选药物与进入临床研究的候选药物之比约为13%,但对于更复杂的中枢神经系统疾病和癌症,该比率可低至0.4%。据估计,将一种新疗法推向市场的研发成本已飙升至高达64亿美元,平均为13亿美元。基础科学研究向临床现实世界影响的转化被贬称为“死亡之谷”。这些挑战已迫使许多制药公司(包括辉瑞、默克、葛兰素史克、阿斯利康和安进)在某个阶段退出神经科学研究,尽管相关疾病是全球致残的主要原因,造成巨大的社会成本,且其中许多疾病无法治愈。
此外,许多被认为是成功的已批准药物,也受到专家和现有证据的批评。相当数量的药物被认为效果甚微,与先前批准的药物相比几乎无额外益处,或仅基于替代终点或有缺陷、证据有限的研究获批。与安慰剂相比,许多已批准药物可能至多只能提供微小的益处,甚至可能降低患者的生活质量和生存率。
更奇怪的是,尽管近几十年来技术大幅进步,药物研发生产力却显著下降。从1950年到2010年,每十亿美元研发支出对应的药物批准数量每9年减少一半,这一趋势被称为反摩尔定律(Eroom's Law)。尽管正如早先预测的那样,这一趋势近年来似乎正在逆转,但诊断药物研发低生产力的主要原因十分重要,因为它阻碍了我们将技术进步和生物医学知识进展转化为改善健康的终极目标。
若干因素被提出是生产力下降的原因:“更易获取和发现的药物”耗尽(低垂的果实);超越先前重磅药物的累积压力(“超越披头士”问题);监管机构风险容忍度降低(“谨慎的监管机构”问题);人力和技术投入增加(“砸钱”倾向);以及高估科技进步(如分子生物学和高通量筛选)的积极影响,这导致仓促放弃先前的方法(“基础研究-蛮力”偏见)。许多人也强调了主流药物发现方法的还原论倾向:基于靶点的药物发现。有趣的是,这种方法大致诞生于这种衰退开始的同时,并且现已主导药物发现方法数十年。大多数基于靶点的药物发现研究本质上是还原论的,因为它们将源于与复杂细胞和细胞外组分网络及其错综复杂反馈环路相互作用而产生的治疗效果,简化为对单个或少数蛋白质行为的调控。尽管如此,需要承认的是,在基于靶点的药物发现中,通过采用多靶点药理学(polypharmacology)方法,已有尝试试图摆脱这种极端还原论。
药物发现始于我们的祖先开始识别所遇物质与其观察到的表型效应之间的关联。例如,桦木孔菌是一种真菌,现已观察到其若干组成物质是有效的免疫调节剂和抗菌剂。证据(包括一具携带此真菌的受感染木乃伊)表明,其在大约5300年前可能已被用于治疗如鞭虫病等传染性疾病。进一步的证据表明,可能有意使用有效药物的历史甚至可以追溯到60000年前。即使现在,临床中使用的一些最关键药物仍可追溯至我们祖先在几个世纪前发现的治疗方法,包括吗啡类似物(21种药物)、阿司匹林、地高辛以及许多其他药物(图4及补充信息1中的“历史上使用”类别)。
经过数千年知识和能力的累积增长,现代生物医学科学的一些先驱,如François Magendie和Claude Bernard,开始揭示这些物质如何对表型产生作用,即其作用机制。随后,Rudolf Buchheim和Oswald Schmiedeberg等开拓性科学家延续此类研究,催生了药理学这一学科(图1)。之后,在20世纪,几位多产的科学家不仅对药理学科学的发展做出了重大贡献,并且将分子如何对表型产生作用的知识应用于设计更好的药物(图1)。

这种方法试图利用现有的科学知识(包括药理学、病理学和生理学方面)来指导和聚焦对随机物质的经验筛选,并将其对表型效应的观察相关联,后来被称为合理药物发现。基于靶点的药物发现可被视为合理药物发现的后续迭代,它与分子生物学革命以及X射线晶体学、核磁共振成像、计算化学、生物技术、DNA测序、组合化学和高通量筛选等技术的发展同时诞生,这些技术使得对生物过程的剖析能够精细到分子与单个蛋白质的结合。
目前,基于靶点的药物发现极大地主导了学术界和制药行业的药物发现方法。针对复杂疾病(如强迫症、抑郁症和阿尔茨海默病)的候选药物,其筛选、选择和优化主要基于它们与一个假设与该疾病发展根本相关的单个蛋白质的结合亲和力。观察分子的治疗效果主要用作最终过滤。
自科学革命和工业革命以来,还原论一直是科学中的主导态度。在20世纪后期和本世纪初期,由于技术进步推动了还原论,这种方法获得了进一步的发展势头。
在还原论主导过去70年的同时,反还原论(甚至被亚里士多德倡导)也在各个领域得到推动。这是由还原论在破解复杂性方面失败所带来的幻灭感所驱动,例如人类基因组计划未实现的承诺。复杂系统科学已经蓬勃发展,并有专门的研究机构。复杂性科学的重要性在2021年诺贝尔物理学奖授予“对我们理解复杂系统的开创性贡献”中得到体现。系统生物学也蓬勃发展,试图利用计算和数学工具更多地关注所研究组分的全面性及其相互作用。在药物发现中,反还原论在几种方法中有所发展,包括系统药理学、网络药理学和多药理学,并在斯温尼和安东尼表明尽管还原论的基于靶点的药物发现占据不成比例的主导地位,但在1999年至2008年间批准的大多数首创药物是通过表型方法发现的之后,获得了额外的动力。这本应开创表型药物发现的复兴;可惜,它仍然因对基于靶点的药物发现的关注而处于边缘地位,并且仅被视为发现新作用机制和首创药物的补充方法。
一个分子对特定蛋白质的亲和力与其对(例如)抑郁障碍的治疗效果之间有什么关系?一个分子与蛋白质的紧密结合能否使人体从如此复杂的疾病中解脱出来?这个问题是针对与基于靶点的药物发现相关的还原论所提出批评的核心。除了许多这些假设的“靶点”甚至可能与其归因的表型无关这一事实之外,大多数疾病和期望的治疗效果似乎过于复杂,无法简化为调控单个蛋白质的行为(也许单基因孟德尔遗传病除外)。此外,已确定的是,我们对许多疾病潜在病理学的知识,在与我们对这些疾病的无知相比时相形见绌。这种知识差距进一步阻碍了在试图抵消特定病理时精确确定单个蛋白质靶点的可行性。
尽管对基于靶点的药物发现提出了许多批评,但缺乏一项全面而系统的研究,能够为以下问题提供相对确定的答案:基于靶点的药物发现是一种高效、最优和合理的方法吗?本文试图通过从以下几个角度审视基于靶点的药物发现方法来提供答案: A. 与还原性较低的方法相比,当前批准的药物中有多少归功于还原论的基于靶点的药物发现?
B. 即使是基于还原论的基于靶点的药物发现所发现的药物,其治疗效果能否简化为对所选蛋白质的结合和调控?
C. 药物以高亲和力与“治疗靶点”的结合与其治疗效果的相关性如何?
D. 基于靶点的药物发现方法是否立足于基于现有科学知识的可靠理论基础之上?
我假设药物发现衰退的一个根本原因是向还原论的基于靶点的药物发现方法的转变,该方法主要根据分子与少数假设相关的“靶点”蛋白质的结合来选择和优化结构,并且通常仅将体内和人体数据用作终端过滤器。另一方面,传统的药物发现必然是一种更经验性的方法,主要依赖于根据分子对人类和其他生物体(以下简称动物)、真菌和细菌的治疗效果来选择和优化分子,因为缺乏还原论所需的工具来直接评估药物在与单个蛋白质相关的较低层级上的效应。
尽管 Swinney 和 Anthony 进行的上述分析在复兴表型药物发现方面具有开创性意义,但他们分析的时期(1999-2008年)非常有限,以至于他们的结论(除了限于建议表型药物发现对于发现首创药物可能更有效之外)后来受到一项评估了更长时间框架的分析的挑战。此外,Swinney 和 Anthony 的分析根本没有涵盖传统药物发现的黄金时期;因此,它不适合比较传统和基于靶点的药物发现的实际贡献。因此,我扩展了Swinney 和 Anthony 的分析,纳入所有批准的药物,并试图提高分析的准确性和客观性。
我手动(非通过自然语言处理等自动化方法)调查了截至2020年底美国FDA批准的所有药物的发现起源
…… 此处省略方法细节描述,感兴趣的读者可以点击文末链接阅读原文~

根据上述治疗类别和发现起源的定义,将“发现起源是观察分子对蛋白质效应”的药物归为基于靶点类,将“发现起源是观察分子对表型效应”的药物归为基于表型类。
总而言之,在分析发现起源时,我尝试通过以下措施最大限度地提高客观性、精确性和准确性: (1) 分析所有药物,而不仅仅是特定时期的药物。 (2) 完全手动分析,而非自动化分析。 (3) 基于对发现起源、治疗类别、基于靶点和基于表型的简单定义做出决策。 (4) 引用原始发现论文和发现者本人的逐字叙述。 (5) 在无法进行(4)的情况下,引用已发表的关于发现过程的文献。 (6) 基于已发表文献和特征树决定治疗类别。 (7) 详细记录识别和分配每个发现起源所遵循的路径。
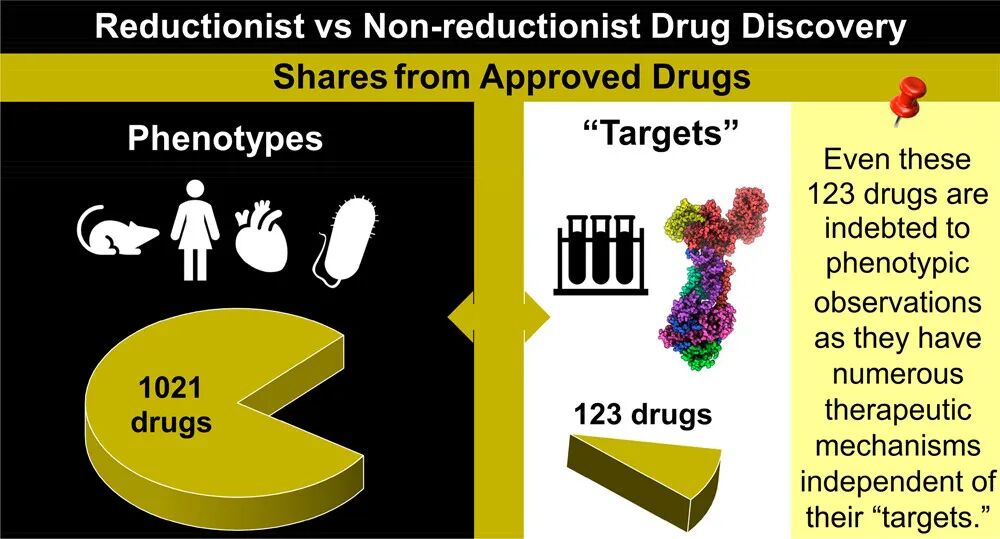
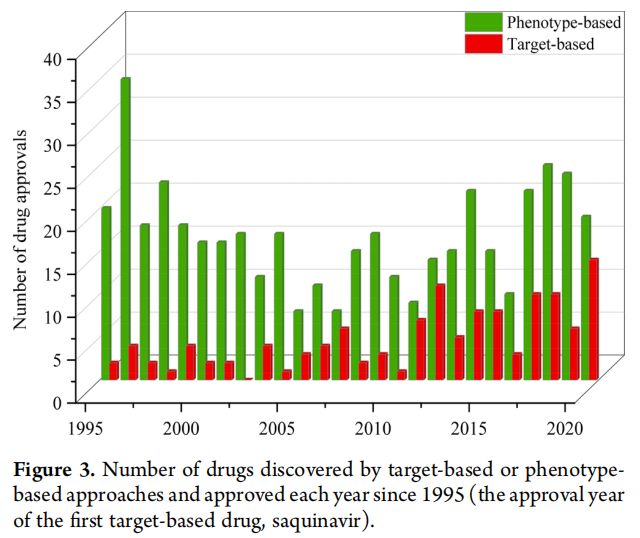
在1310种美国FDA批准的药物中,69种是基于内源性生物药,97种是其他生物药。在剩余的1144种小分子药物中,123种(10.75%)通过基于靶点的药物发现发现,1021种(89.25%)通过基于表型的方法发现。尽管小分子基于靶点的药物发现在过去40年中占主导地位,但它在当前使用的药物中所占份额微乎其微(123种药物,占9.39%),而与基于表型的方法(1021种,占77.94%)形成对比(图2a)。即使只考虑1995年以后批准的药物,这种不成比例的差异仍然存在(基于靶点:123种,占17.30% vs 基于表型:438种,占61.60%)(图2b)。尽管每年批准的药物中基于靶点发现的份额似乎随时间增长,但没有一年其份额不超过表型药物发现的份额(图3)。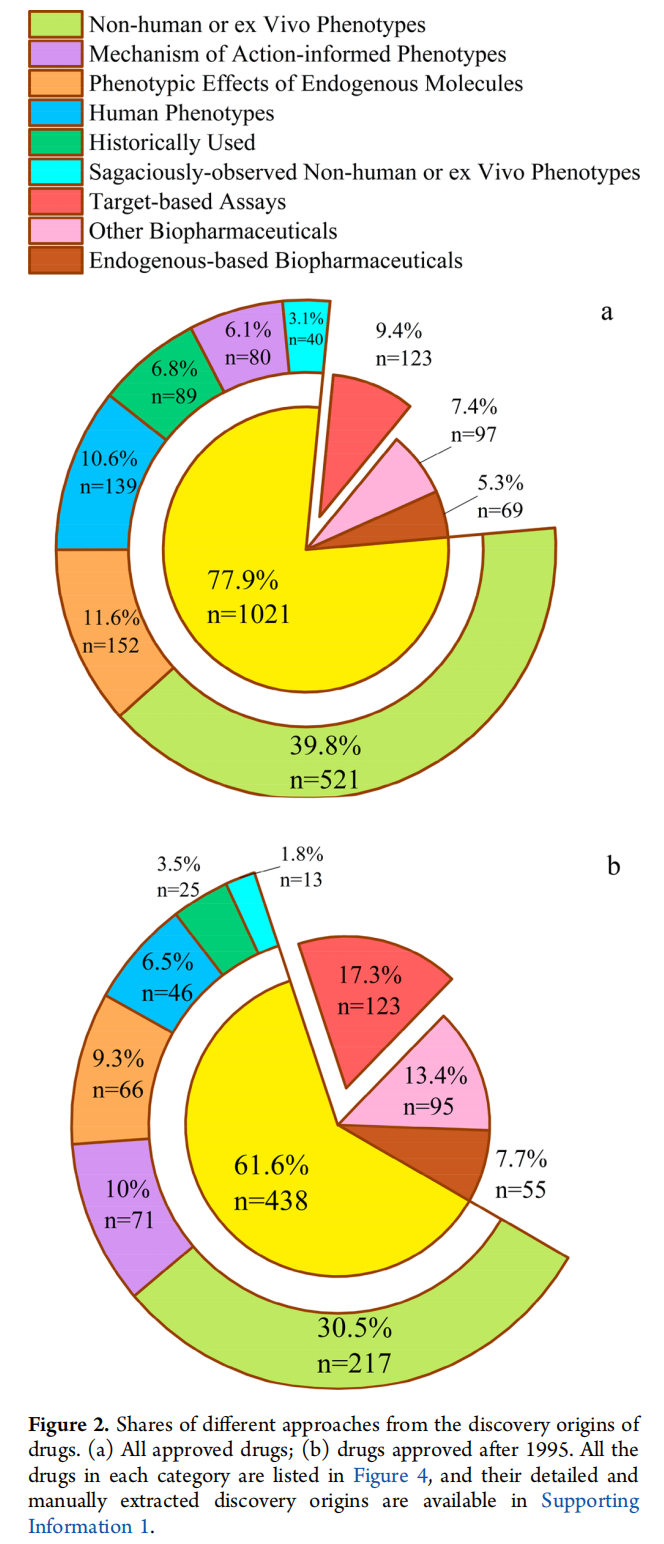
回顾补充信息1中提供的发现起源表明,与过去几十年作为标准的教科书式药物发现程序相反,临床使用的许多最重要药物中,相当一部分起源于许多当今药物探索者可能认为是“超乎寻常”和“发生在另一个星球”的方法。大多数已批准的药物源于强调和使用高度预测性的表型模型,例如预测性动物模型、体外系统或细菌、真菌和原虫培养物(约700种药物)。这一点在许多药物的发现起源中显而易见。
对模型预测性的关注如此之高,以至于在许多情况下,药物探索者将自己作为模型并对分子进行自我实验。基于偶然性和敏锐观察非人类或体外表型发现的药物也可以被认为归功于这种对预测模型的关注。此类偶然发现在基于靶点的药物发现的生化筛选中是不可能的。
敏锐的临床医生观察到的表型效应也贡献了大量药物。例子包括氯丙嗪(几乎所有抗精神病药的原型)、丙咪嗪和异烟肼(几乎所有抗抑郁药的原型)以及对乙酰氨基酚、皮质类固醇、双硫仑、几种利尿和糖尿病药物、甲基多巴、米诺地尔、氟班色林、吉非贝齐、纳比隆、阿那格雷、西地那非、壬二酸、对苯二酚和美金刚。公平地说,还应加入许多基于历史观察发现的药物。这些案例凸显了临床环境中敏锐观察的重要性。
药物的发现起源表明,自上世纪以来药物发现演变过程中发生显著转变的另一个方面是药物发现团队的结构和功能。几十年前,药物发现团队规模小,由少数药物化学家和药理学家组成,所有人都试图回答同一个问题,并直接监督化学结构、修饰及其对表型效应之间的关系。如今,药物发现团队规模显著更大,发现药物的目标已被简化和专业化为不连贯的任务。上世纪药物发现团队的不过度专业化的结构,使得表型观察能够无缝地、反复地转化为结构选择和优化,并观察这些优化的表型结果。另一方面,当前药物发现团队的专业化和碎片化结构,更适合还原论的基于靶点的药物发现方案:单向的串行过滤和漏斗式流程,其中表型观察主要仅用作终端过滤器。
这一见解可以从收集的数据中推断,即“近期生产力的提高”更多可追溯至制药行业对其失败的适应,而非解决其根本原因。例如,努力已从未解决的复杂中枢神经系统疾病挑战,转向更可简化的罕见病和单基因疾病,或衍生几十年前发现的药物的类似物。甚至在有些情况下,几十年前未推向市场的药物被重新启用,并促进了表面上的“生产力提高”。这些回归几十年前结构和分子的案例的奇特之处,可以通过注意到类药化学空间估计是如此巨大(高达10^60个分子),以至于新结构本不应稀缺来阐明。
还应公平地补充,表型观察的不成比例贡献还包括许多批准药物增加的批准后标签内和超说明书适应症。在许多情况下,此类重新定位和新增适应症是基于敏锐的临床医生进行的表型观察。
尽管我们发现目前使用的大多数药物是通过如今已不那么普遍的非还原论方法发现的,但这并非还原论式的靶向药物发现方法效率低下的决定性证据,因为如前所述,这种差异可能由其他因素造成。
我们可以从另一个角度评估靶向药物发现:质疑其在那些被我们归类为“靶向”来源的药物发现中的作用。这有助于探究此类药物的治疗效果是否真的可归结为对单一蛋白质的调节。我们知道,大多数药物因缺乏疗效而无法通过临床阶段,尽管其中许多是基于靶向药物发现方法发现的,并且能以高亲和力结合其靶点。这意味着,除了高亲和力结合靶点之外,可能还存在其他影响药物有效性的变量。其中一个变量是所选靶点与疾病的相关性程度。有趣的是,研究表明,许多假定的靶点与疾病之间并无可靠关联。另一种可能性是,药物的治疗效果可能源于对靶点之外众多组分的恰当调控。药理学中公认,许多药物的治疗效果是由多种机制介导的(见补充信息3)。此外,最近研究表明,“脱靶毒性是正在进行临床试验的癌症药物的常见作用机制”。考虑到这些事实,以及即使在靶向药物发现中,治疗效果评估也作为终端过滤器发挥着一定作用,我们可以假设:即使对于“靶向”药物,其治疗效果也可能不完全归因于靶向药物发现方案。治疗性的“脱靶”机制可能被表型终端过滤器无意识地、偶然地筛选出来。
这一假设通过以下观察得到加强:当代临床实践中使用的许多获批药物,其发现起源相当朴素。它们是通过在人体或体内模型中观察其效应而被发现的,或是偶然发现,或是仅筛选了数百个分子(补充信息1),而非通过系统性筛选大型化合物库。
换句话说,将我们在上一节中确定的“靶向”药物的发现归功于靶向药物发现及其对单一“靶点”的亲和力,可以视为幸存者偏差的一个案例。幸存者偏差是一种逻辑谬误,即只关注那些成功通过某个筛选步骤的实体,而忽视那些未通过的实体(通常因为它们缺乏可见性)。当大多数为高亲和力结合单一“靶点”而设计的药物在临床试验的疗效阶段失败时,那些碰巧通过测试的药物的疗效,不应简单地归因于其高结合亲和力。
为验证“许多通过‘靶向’方法发现的药物的治疗效果得益于‘脱靶’机制”这一假设,我对上一节确定的每一种“靶向”药物进行了系统评价(遵循循证医学原则)。我在PubMed和Embase中使用了以下查询策略:PubMed为“[Drug]/pharmacology[Majr]”,Embase为“Drug/exp/mj/dd_pd”。检索到的引文使用系统评价助手-去重模块进行去重(各药物的检索结果及去重后总数见补充信息2)。我手动(非自然语言处理等自动化方法)审阅了这些引文,并提取了那些经实验证明或提示其介导了药物最初获批适应症的治疗效果、但并非由该药物发现所依据的“靶点”所介导的作用机制。
为排除药物结合其“靶点”所产生的下游效应,我从提取的“脱靶”机制中排除了那些由第一层或第二层相互作用蛋白质所介导的机制。我从STRING v11中获取了这些相互作用蛋白,设置如下:主动交互来源为“实验”或“数据库”;最低交互得分要求为最高置信度(0.900);第一和第二层最大交互蛋白数为500。那些基于药物直接结合“脱靶”蛋白的“脱靶”机制不受此排除标准限制。由于在许多情况下,多项不同研究提及了同一药物治疗效果的相同机制,我记录并引用了所有这些不同研究,但仅在表格中突出显示其中一个重复项。表2中的计数与补充信息3中为每种药物列出的机制数量不同,原因是表2中的计数反映了去重后的机制数量,这些机制也列于补充信息3各药物的表格前。
总结来说,提取的机制需满足以下标准:
基于所审阅的实验药理学研究,这些机制被提出在某种程度上介导了药物获批适应症的治疗效果。 这些机制既不依赖于药物发现所依据的“靶点”,也不依赖于这些“靶点”的第一或第二层相互作用蛋白,除非该机制是基于药物直接结合这些第一或第二层相互作用蛋白。
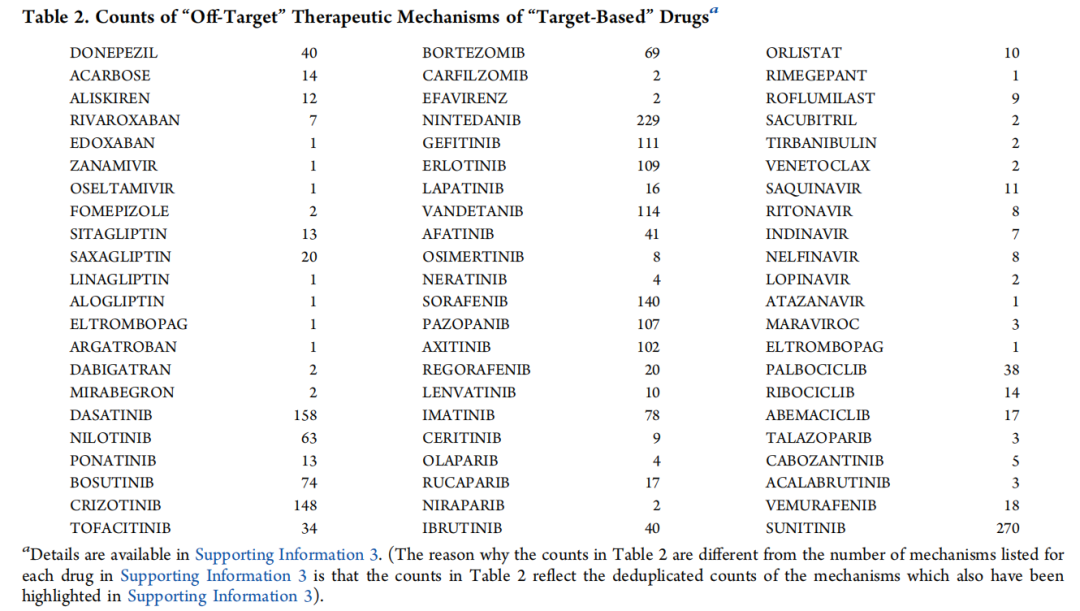
系统评价(包括对31027篇独特文章的手动审阅)证实了该假设。许多“靶向”药物具有大量“脱靶”治疗机制(表2及补充信息3)。例如,多奈哌齐是作为乙酰胆碱酯酶抑制剂被发现和开发的,但研究发现其拥有40种不依赖于乙酰胆碱酯酶的治疗机制,包括抗炎和免疫调节作用;血管保护、神经保护和抗氧化特性;抑制神经元凋亡;通过脑源性神经营养因子、胰岛素样生长因子1和SRC等多种途径增加神经发生;刺激少突胶质细胞分化和髓鞘相关基因表达;对微管亲和力调节激酶4、烟碱型乙酰胆碱受体、σ非阿片细胞内受体1以及钙、钾、钠通道的直接作用;下调miRNA-206;防止谷氨酸神经毒性;对抗氧-葡萄糖剥夺诱导的损伤;调节血清脂肪因子水平;增加抗β淀粉样蛋白自身抗体的表达;增加α-分泌酶活性并降低β-分泌酶活性;减少β淀粉样蛋白的产生并增加其清除;减少tau蛋白磷酸化;以及增加ADAM10和ADAM17的运输和活性。
另一个例子是克唑替尼。除了抑制MET和ALK外,它能以相当高的亲和力结合其他146种激酶,诱导氧化DNA损伤和细胞凋亡,并通过抑制巨噬细胞刺激1受体和上调主要组织相容性复合物分子产生免疫调节作用。
这意味着,靶向药物发现对获批药物的贡献甚至远低于9.4%。如果完全依赖于还原论的靶向药物发现方案,这些“脱靶”治疗机制都不会存在。它们是因为治疗效果的终端评估而被无意识和盲目地筛选出来的,并且不一定伴随所有基于结合单一“靶点”而筛选出的分子。值得注意的是,这些计数 不可避免地 仅限于迄今已阐明的机制;可以合理预期,实际的“数量”会高得多。
既然我们已经观察到大多数药物是通过表型观察发现的,甚至“靶向”药物也不像宣称的那样“靶向”,那么,作为靶向药物发现“圣杯”的高亲和力结合“靶点”,对于药物的治疗效果究竟有多大相关性?通过筛选和过滤对“靶点”具有更高结合亲和力的分子,靶向药物发现基于这样一个前提:具有治疗效果的药物在对“靶点”具有高结合亲和力的分子中富集。这一前提的一个粗略推论是,已获批的药物对其治疗效果介导“靶点”可能具有相对较高的结合亲和力,至少当其作用机制基于竞争性拮抗和抑制时是如此。
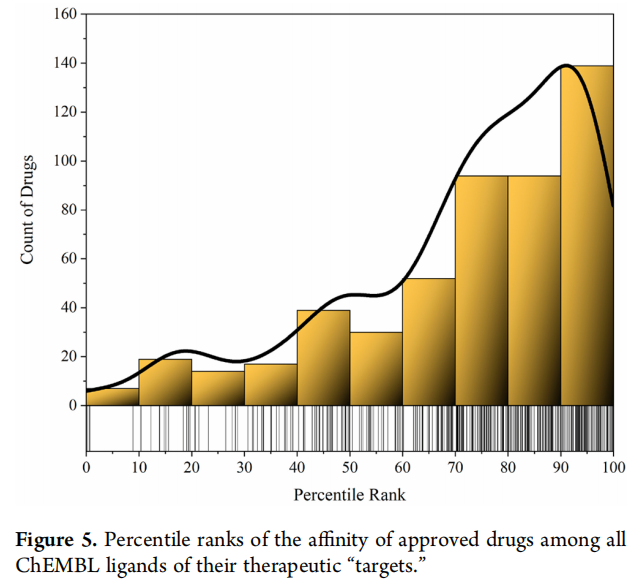
为验证靶向药物发现的这一前提(即具有治疗效果的药物在其治疗“靶点”的所有配体中,高亲和力分子里富集),我考察了每个获批药物对其各治疗“靶点”的结合亲和力,在ChEMBL中该“靶点”所有配体的亲和力中的百分位等级。我从“分子药物靶点综合图谱”中获取了获批药物的靶点,该图谱汇编了获批药物的“治疗靶点”,定义为“药物直接结合并负责其治疗效果的蛋白质或其他生物分子(如DNA、RNA、肝素、多肽)”。我排除了具有以下作用机制的药物-靶点对:激动剂、激活剂、生物药物、通道开放剂、调节剂、变构拮抗剂、部分激动剂、反向激动剂、DNA和RNA抑制剂、“细胞膜抑制剂”、释放剂和螯合剂。然后,我从ChEMBL中获取了剩余“靶点”可用的结合测量数据,并进行以下整理:排除缺乏pChEMBL值或pChEMBL值关系非“等于”(如“小于”或“大于”)的测量值;排除非IC50或Ki值的测量值;基于先前关于抑制活性测量值可比性的研究,对以IC50值表示的测量值,其pChEMBL值加上0.30以增强可比性;对具有多个有效测量值的分子,取其pChEMBL的平均值,记录每个配体的最终pChEMBL值。排除剩余配体数少于100的“靶点”后,我计算了获批药物的亲和力在剩余配体中对每个靶点的百分位等级。每种药物的所有盐形式和质子化替代形式的百分位等级取平均值,并根据每种形式的测量值计数进行加权。
尽管相当数量的获批药物对其治疗“靶点”的亲和力相对较低,但大多数倾向于处于最高的百分位等级(图5)。(所有药物-靶点对的详细数据见补充信息4,汇总数据见补充信息5。)这表明,高亲和力结合“靶点”与治疗效果相关。结合靶向药物发现对获批药物的微小贡献以及“靶向”药物众多的“脱靶”治疗机制来看,可以认为,高亲和力结合单一“治疗靶点”只是药物对表型产生治疗效果的一个方面。
通过评估其理论基础的稳固性,我们可以就靶向药物发现的效率和合理性得出更明确的结论。
在几年前我听的一场关于药物发现与设计的介绍性讲座中,人体被比作一个时钟,疾病被比作时钟中一个齿轮的缺陷,而药物发现的目的被阐述为设计一个能够结合这个缺陷齿轮的分子。这个比喻揭示了靶向药物发现的一个深层次谬误:假定人体的功能可与机器相提并论。这种对生物系统的机械论思维是一种还原论谬误,已盛行数百年。它源于我们设计和构建系统的人为方法。当我们旨在设计和构建一个特定系统时,我们会为每个部件设计其在整体系统最终功能中的特定作用。因此,汽车或宇宙飞船的每一项功能或功能障碍都可以通过一系列任务链追溯到特定部件。
机械论思维的谬误在于其预设了这种部件与功能之间清晰的任务链在所有系统中具有普适性。生物系统并非采用同样方法设计的结果。它们高度复杂,是经过选择进化的产物。选择并不为部件分配特定角色。它只是在随机变异产生的系统各种最终输出和表型中进行选择。
所有生物体,与城市、互联网和股票市场类似,都是自组织复杂系统。它们的部件并不与其行为绑定。尽管某些元素在某些功能或功能障碍中可能作用突出,但通常没有清晰可分的界限。这导致复杂系统相比链式系统具有几个普遍特征:不同部件具有广泛的多功能性;部件状态与系统最终行为之间存在连续且谱状的因果关系,而非清晰、二元式的;通过个体部件与整个系统之间频繁且广泛的反馈实现“自我控制”的能力;广泛冗余;残留性;以及简并性或多重实现性,即多组不同的过程可产生相同的最终输出。
因此,靶向药物发现理论框架的一个核心问题是:它遵循机械论思维的谬误,忽视了复杂系统与链式系统之间的区别,对生物过程的组织和功能采取了过于简单化的态度。关于生物系统复杂性含义的数学讨论,以及“当存在非严格线性因果链时,对网络单一节点施加刺激不可能对系统整体行为产生实质性改变”的论述,见参考文献147。
我们已经提供了大量证据支持表型药物发现比还原论靶向药物发现更高效的观点。但这是为什么呢?仅仅是因为许多疾病无法归结为单一蛋白质吗?如果这样,基于多靶点的药物发现(如多药理方法)也能同样高效吗?
幸运的是,整合信息理论为此提供了一个数学化和形式化的框架来进行研究。尽管这个形式化理论(其预测已得到经验和数学证据的证实)正在研究中,并且其解释意识的有效性受到一些批评,但其从系统“内在视角”研究各种系统因果结构的适用性已得到充分证明。简而言之,IIT(integrated information theory)研究“系统的各个部分如何通过处于特定状态来约束系统自身潜在的过去和未来状态”。
基于IIT,研究表明,在时空粗粒化的更高层次上描述系统状态,可以增加信息和内在因果效应力(Φ)(图6)。这种增加主要是因为,当嘈杂(非确定性)和简并的微观元素被分组为更确定、简并性更少的高层次描述时,高层次描述能捕获更具体的系统机制。后来,在一个生物学模型中得到证实,与基于平均的粗粒化相比,通过黑箱化,时空高层次描述能提供甚至更强的内在因果效应力,尤其是在具有异质性、整合的专门部件的系统中(以生物体为典型)。黑箱化最初由控制论提出,通过隐藏内部运作、仅考虑输入和输出来实现对高度复杂系统的建模。应该指出的是,正如控制论奠基人之一Ross Ashby所阐述的:“然而,黑箱理论的应用比这些专业研究更广泛。试图开门的孩子必须操纵把手(输入)以在门闩上产生期望的运动(输出);他必须在看不到连接它们的内部机制的情况下,学会如何通过控制前者来控制后者。在日常生活中,我们处处面临内部机制无法完全被审视的系统,必须用适用于黑箱的方法来对待。”可以说,粗粒化和黑箱化通过捕获高度复杂的低层次中更简单的高层次吸引子状态和参数空间压缩,增加了因果效应力和信息量。参考文献403-416提供了其他一些在更高层次上增加简洁性和信息量的例子。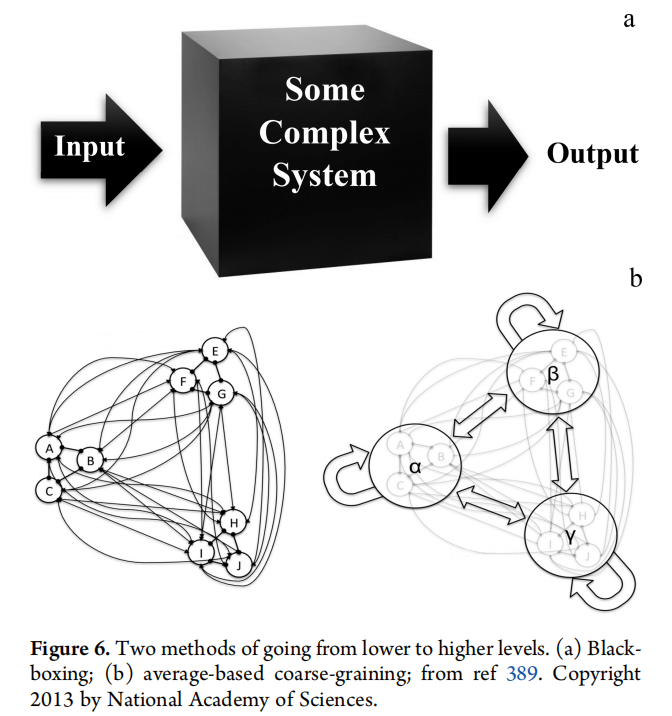
所有这些意味着,即使靶向药物发现记录了分子对人体所有蛋白质及每一个其他组分的影响,如同一个假设的、不切实际的理想多药理靶向方案,其效率仍将低于表型药物发现。因为表型药物发现在更高层次上(黑箱化)研究分子对人体的影响,故而具有卓越的因果效应力和信息量。这一点在药物发现中使用的生成式和预测性机器学习模型中,关于“最好提出什么问题”和“最好靶向哪个变量”的背景下尤为关键。
在药物发现史上,各种技术,如计算化学、高通量筛选和组合化学,都曾崭露头角,最初获得极大关注和乐观期待,但随着时间的推移,其局限性也逐渐显现。如今,这些工具已成为许多药物研发活动中不可或缺的组成部分,但人们对它们的期望已趋于现实。本研究分析的数据表明,该领域需要对靶向药物发现的局限性有类似的认识。
然而,必须承认,过去几十年开发的大量靶向工具在未来道路上能提供巨大帮助。首先,一些疾病,如单基因孟德尔遗传病,可在不同程度上归结为单一或少数几个蛋白质,因而适合采用靶向药物发现方法。一些单克隆抗体的成功也说明了将某些疾病归结为少数几个蛋白质的可行性。其次,在基于类似物的药物发现背景下,靶向药物发现是修饰和优化结构的重要利器。基于类似物的药物发现本身就是一个有价值的工具,可在药物治疗中获得渐进式但仍重要的改进。此外,对于某些疾病,收集足够且合适的更高层次数据可能效率太低、负担过重。在这些负担中,即使类器官、芯片器官和计算机模拟试验等技术可以减轻需求,但可能仍需牺牲大量动物这一点值得注意。在这些情况下,靶向工具可以发挥重要作用。靶向工具的另一个重要作用是分析治疗效果的低层次基础。从靶向方法获得的低层次数据可用于系统生物学、系统药理学和多尺度建模等方法,以帮助研究治疗效果如何从低层次现象中涌现。当这些分析被恰当地整合到试图从低层次和高层次数据中获益的药物发现框架(如下文建议的框架)中时,它们将具有巨大价值。
基于我们所分析的大量数据,当前药物发现中占主导地位的还原论方法似乎是低效的。相反,在选择和优化分子时优先考虑更高层次的观察,似乎是一种基于证据的提高效率的方法。我们已经看到,合理药物发现源于试图根据现有科学证据来指导和聚焦对随机物质的经验筛选。从这个意义上说,还原论的基于靶点的药物发现是不合理的,因为现有的科学证据似乎与其方法论相矛盾。从合理药物发现向还原论的基于靶点药物发现转变过程中的关键问题和偏差,在于所采用的还原论程度。利用药物作用机制知识的合理药物发现,与还原论的基于靶点的药物发现试图将药物的治疗效果简化为单一"靶点"蛋白质,二者之间存在巨大差异。
重要的是,通常治疗效果不能简化为一个分子与少数几个蛋白质的结合。这种不可简化性在1.6.2节和附录信息3中已阐明,我们发现许多有靶点起源的获批药物,其治疗效果得益于众多的脱靶机制。通过黑箱化人体及其疾病的巨大复杂性,表型药物发现可以规避人体内部的所有复杂性,直接瞄准最终目标——即治疗效果本身,而非该目标的一个假设的低层次组成部分。这种更直接、更紧密的联系能提供更高的预测性,这本身对(药物)批准率有显著影响。
可能有人会反驳我的批评,认为基于靶点的药物发现在不同程度上利用了在动物和人体中进行表型观察的益处。确实如此,这也正是我们在1.6.2节中进行研究的基础。然而,如前所述,问题在于基于靶点的药物发现中的表型观察并非主要用来指导结构设计,因为它们仅被用作单向串联漏斗式筛选中的最终过滤器。分子的结构首先是根据其与靶点的结合亲和力来设计的,仅在最后步骤才根据其治疗效果进行筛选。相反,在表型药物发现中,分子是通过直接观察所有分子的表型效应来筛选的。正如1.6.2节和附录信息3所证明的,分子与单一蛋白质的结合只是治疗效果的一个组成部分,高亲和力结合单一靶点与引发治疗效果之间可能相关性太弱。
更高层次观察所能提供的增强的预测性,对于人工智能和机器学习在药物发现中的应用具有重要意义。现有数据表明,这些强大的技术目前通常未被用于回答正确的问题。最终目标是设计和选择对表型具有期望治疗效果的分子,因此提出的问题需要与这一目标相称。然而,在基于靶点的药物发现框架下,这个目标被分割和简化为一些附属目标,例如设计对单一"靶点"具有更高亲和力的分子。根据本文呈现的证据,这些附属目标可能与最终目标关系不大。基于我们在1.6.4节的讨论,根据治疗效果本身(而非这些效果的单一组成部分)来选择和优化分子,能够提供远更强的因果效应力。除了在临床研究中成功证明疗效和获得上市批准的比率之外,这还能极大地影响(获批药物的)效应大小、临床意义和现实世界价值;这些获批药物的重要方面,正如我们在文章前面讨论过的,已成为专家和现有证据批评的对象。
一个非常初步的设想是,可以开发一些机器学习模型,其目标变量是分子在人体中或某些高度预测性模型里的治疗效果的度量。这些模型的输入特征可以是药物对较低层次效应的各种变量,例如,借鉴系统综述等方法,挖掘已发表文献中可用的动物和人体数据(以及未发表的数据,如电子健康数据、患者报告结果和专有数据)。这些以数百万生命、数十亿美元和数十年研究为代价获得的巨大数据宝藏,可以通过系统综述等方法进行挖掘和利用。当然,这个框架非常初步,并且在许多情况下,收集足够数量和可接受质量的低层次和高层次数据来构建体面的机器学习模型可能不切实际。尽管如此,这种方法(或可称为"基于化学信息学的荟萃分析的系统综述")即使在这些情况下也能提供有价值的初步定性见解。
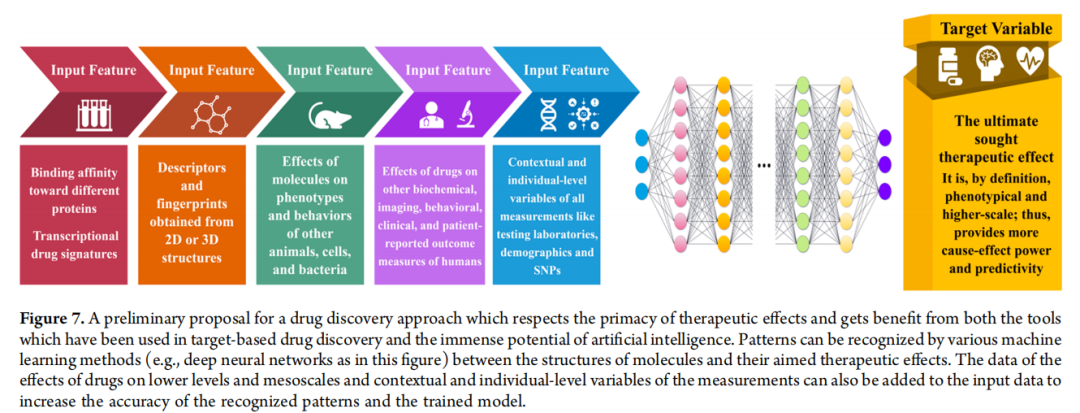
我们可以期望,在认识到基于靶点的药物发现的局限性,并装备其现有能力以及机器学习和深度学习、高内涵成像、高通量表型分析、可穿戴生物传感器、物联网、类器官和电子健康数据等先进技术,同时更加关注概念性进展之后,我们不仅能够超越当前的低生产力,甚至能够用更安全、更有效的替代品来取代当前的金标准药物。
新兴的基于靶点的方法(如蛋白质降解技术)未来有潜力缓解基于靶点的药物发现整体效率低下的问题;然而,由于它们最近才出现,未能在本分析中得以体现。
关于1.6.1节,必须指出目前尚无确定的标准和定义来判定哪些药物应归类为基于靶点的或基于表型的。因此,本文提供的定义可能会引起争议。鉴于缺乏预先确立的定义,本分析中使用的定义不可避免地带有主观性。尽管如此,我已尽力提供简单的定义,力求务实、对本研究目的最有用,并在分类中提供最大的客观性。这些标准也与 Swinney 和 Anthony 的开创性研究中采用的标准一致。此外,由于药物发现的过程高度复杂且每种获批药物都具有独特性,试图将其归入离散且简明的类别可能会在定义和分配类别时引入主观性。我试图通过充分记录我识别和分配每个发现起源所遵循的路径来减轻这种二元偏倚。
我未能找到几种药物的发现起源。在某些情况下,发现起源无法明确识别,且没有记录是哪项观察首次确定了某个治疗类别与其治疗效果之间的关联。对于那些未找到准确发现过程记载、但根据其发现年份和当时的趋势无疑是基于表型观察发现的药物,其发现起源被归类为非人类或离体表型观察。存在这样的情况:我找到了与发现相关的论文(例如在 PubMed 上),但无法找到其全文网页。由于特征树与生物学无关,为定义类似物设定一个静态的截断值是不可行的。
关于1.6.2节,生物体行为的机制很难定位。即使是使用 siRNA 等最先进的方法来阐明药物作用机制也存在诸多缺陷,使用药理学工具化合物进行动物研究以调查药物机制也是如此。我在调查"基于靶点"药物的"脱靶"治疗机制时使用的许多研究就属于此类。我所统计的一些"脱靶"机制与药物治疗效果的相关性是基于普遍关联而非明确证据,尤其是那些源自激酶组抑制试验的机制。此外,尽管我已尽力,但一些被计为独立治疗机制的机制仍有可能追溯至相同的根本机制。
至于1.6.3节,虽然 ChEMBL 汇编了每种蛋白质的配体而未基于低或高亲和力施加限制,但可以合理假设,从 ChEMBL 中检索到的每种蛋白质的配体,与那些尚未测量其对该蛋白质亲和力的随机选择的配体相比,富含具有更高亲和力的分子。因此,可以合理假设,获批药物的亲和力在随机分子中的百分位等级会高于在每种蛋白质的 ChEMBL 配体中的百分位等级。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢