
肿瘤免疫微环境(TIME)在癌症进展及免疫治疗响应中发挥着关键作用。多重免疫荧光(mIF)是一种解析TIME的重要成像技术,但其高昂成本、流程复杂以及通量受限,严重制约了其在大规模人群研究和临床实践中的广泛应用。
针对这一关键瓶颈,微软研究院、华盛顿大学与Providence Genomics等机构的研究团队于2025年12月9日在《Cell》上联合发表了题为“Multimodal AI generates virtual population for tumor microenvironment modeling”的研究工作,其中微软研究院的Hoifung Poon、华盛顿大学的王晟以及Providence癌症研究所的Carlo Bifulco共同担任通讯作者。

研究提出了一种极具前景的多模态人工智能框架GigaTIME。该方法通过将常规获取的H&E图像转化为高度信息化的虚拟mIF图像,实现肿瘤免疫微环境的规模化建模,为人群尺度的TIME分析与发现推动精准免疫肿瘤学的发展奠定了基础。
GigaTIME 代码仓库:
https://aka.ms/gigatime_code
背景
肿瘤免疫微环境(TIME)在癌症进展中发挥着关键作用。它通过影响肿瘤的免疫监视与免疫逃逸,进而调控肿瘤生长、侵袭、转移以及对癌症治疗的反应。TIME是一个高度复杂的空间生态系统,由癌细胞以及多种非恶性细胞类型构成。免疫组织化学(IHC)能够可视化特定蛋白的激活状态,是揭示TIME中关键细胞状态的重要工具。IHC的一个关键局限在于,单次实验通常只能检测一种蛋白,且不同蛋白往往需要在不同的组织切片上分别评估。这一限制在肿瘤微环境建模中尤为突出,因为理解肿瘤细胞与多种免疫细胞之间复杂而动态的相互作用,往往依赖于对多种蛋白信号的同时观测。
多重免疫荧光(mIF)作为一种有力的替代技术应运而生,它能够在同一组织切片上实现多通道蛋白的共定位分析,同时保留组织的空间结构。尽管前景广阔,mIF在大规模研究中的应用仍然受到显著限制。这主要源于其高昂的成本,包括试剂、专用设备及计算基础设施的投入,同时其染色、成像和数据处理流程高度依赖人工操作。由此导致现有mIF数据资源极为稀缺,严重制约了其在大规模临床发现与转化研究中的应用潜力。
相比之下,苏木精-伊红(H&E)图像在临床流程中可低成本、常规化地获取,广泛用于研究组织结构和细胞形态。尽管H&E图像不能直接揭示细胞状态,但其所呈现的细胞空间分布模式可以为推断细胞个体状态提供线索。这类模式对人类专家而言可能并不直观,却有望被先进的多模态人工智能模型系统性地挖掘和利用。近年来,基础模型的发展进一步放大了这一潜力,通过在大规模病理图像数据集上的预训练,人工智能模型已展现出卓越的表征学习与泛化能力。这些进展共同表明,从H&E图像中学习能够指示空间分辨蛋白激活状态的病理特征,在技术上是可行的,也为TIME的规模化建模提供了全新的可能性。
结果
GigaTIME生成多重免疫荧光的虚拟人群
作者首先通过实验获取了441张mIF图像,这些图像来源于21张H&E染色切片,覆盖21个蛋白通道(表1)。随后,这些配对的H&E与mIF切片通过一套计算流程进行处理,包括图像配准与细胞分割,最终构建了一个包含4000万个细胞的配对H&E–mIF数据集(图1A)。
表1 本研究中使用的TIME标志物及其细胞表达情况
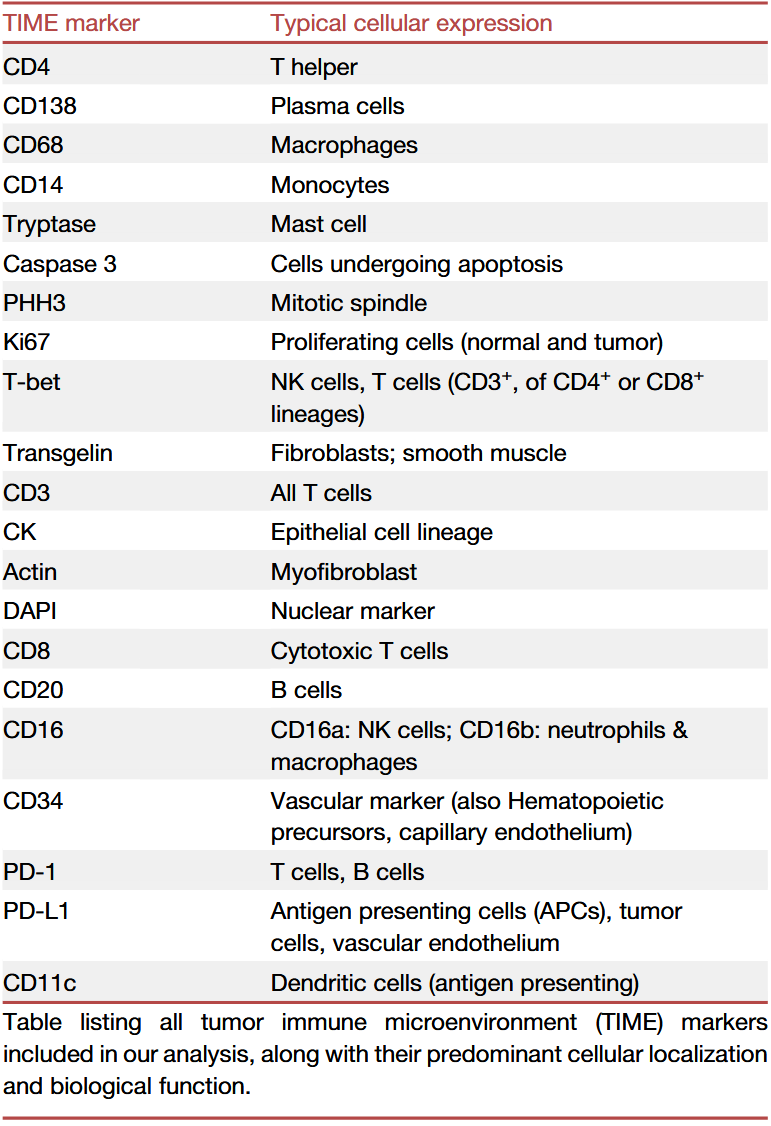
将配对数据划分为训练集、开发集和独立留出的测试集。为实现从H&E图像到mIF图像的转换,GigaTIME在训练集上进行训练,采用基于NestedUNet的分块式编码器–解码器架构。模型以H&E图像块作为输入,输出21个对应的mIF图像块,每个块对应一个蛋白通道。随后,将这些通道特异性的图像块拼接重建为整张mIF全切片图像,从而实现具有空间分辨率的切片级蛋白激活谱分析。具体而言,对于给定的蛋白通道,GigaTIME会为每个像素输出一个二分类标签,指示该像素在该蛋白通道下是否处于激活状态。基于此,可以统计任意图像块或整张切片中被激活像素的数量,以及激活密度得分。
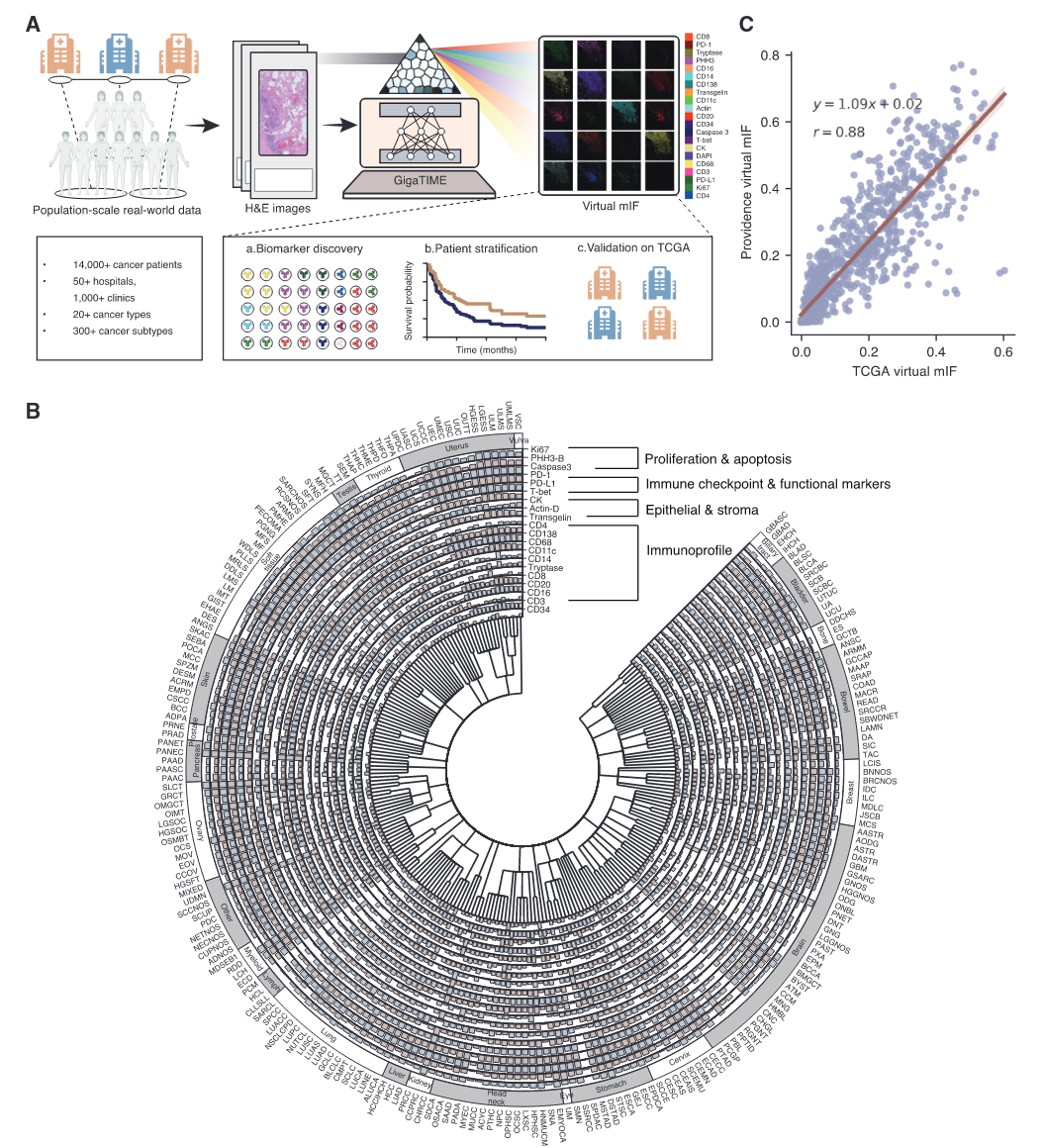
图1 GigaTIME实现人群尺度的肿瘤免疫微环境分析
随后,将GigaTIME应用于一个大规模且多样化的真实世界数据集,该数据集包含Providence Health体系中来自美国七个州、51家医院和1000余家诊所的14256张H&E全切片图像,覆盖24种癌症类型和306个癌症亚型。利用训练好的模型,为这些患者生成了299376张虚拟mIF全切片图像。由此,作者构建了一个大规模、多模态的虚拟人群,包含H&E图像、虚拟mIF图像以及生物标志物、分期和生存状态等临床属性。作为概念验证,为每张mIF图像计算了蛋白激活密度得分,定义为激活像素的比例。随后,通过对同一癌症亚型的肿瘤进行均值汇聚,获得了基于mIF的TIME特征谱,覆盖不同癌症亚型(图1B)。为评估该方法的稳健性,研究团队进一步将GigaTIME应用于TCGA的10200例肿瘤样本,生成了214200张覆盖21个通道的虚拟mIF全切片图像。结果显示,基于Providence和TCGA两个虚拟人群所得到的聚合激活得分具有高度一致性(图1C),突显了GigaTIME的泛化能力与可靠性。
GigaTIME将H&E全切片图像转换为mIF图像
作者将GigaTIME与常用于虚拟染色任务的CycleGAN模型在不同粒度层级(像素级、细胞级和切片级)上进行比较。结果显示,GigaTIME在21个蛋白通道中的15个上显著优于CycleGAN,其余6个通道未观察到统计学显著差异(图2A)。在细胞级评估中,GigaTIME的相关性显著高于CycleGAN,而后者的表现接近随机水平,表明CycleGAN未能恢复连贯的细胞级模式(图2B)。为评估全局空间模式,作者实现了一种受免疫评分启发的切片级指标。GigaTIME在DAPI通道上的Spearman相关系数达到0.98,在所有通道上的平均相关系数为0.56;而CycleGAN在所有通道上均接近零相关(图2C)。最后,对具有代表性的全切片图像块进行的定性比较,进一步直观展示了实测mIF与GigaTIME转换得到的虚拟mIF之间的高度一致性(图2D)。
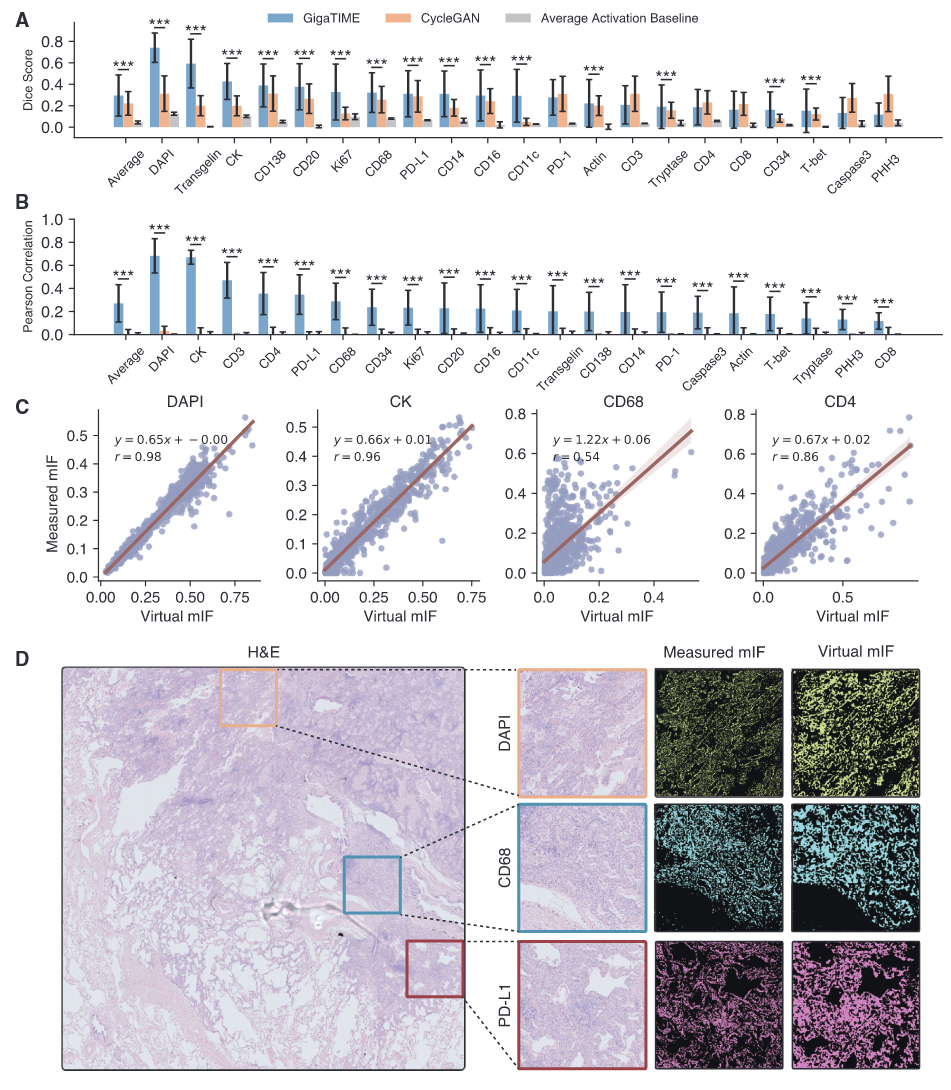
图2 GigaTIME实现从H&E到mIF图像的转换
虚拟人群支持蛋白–生物标志物关联的大规模发现
研究团队构建的虚拟人群在泛癌、癌种以及癌症亚型三个层面,识别出了21个由GigaTIME转换得到的虚拟蛋白通道与20个临床生物标志物之间的1234项具有统计学显著性的关联(图3A)。具体来说,在泛癌层面,共识别出175项显著的蛋白–生物标志物关联(图3B),其中许多结果得到了既有文献的支持。在癌种层面,GigaTIME在脑肿瘤中识别出64项蛋白–生物标志物关联(图3C),在肺癌中识别出137项(图3D),在肠道肿瘤中识别出175项(图3E)。上述关联中有相当一部分具有明显的癌种特异性。在癌症亚型层面,虚拟人群揭示了许多组织学特异性的关联,而这些关联在样本量较小的队列中往往难以发现(图3F和图3G)。
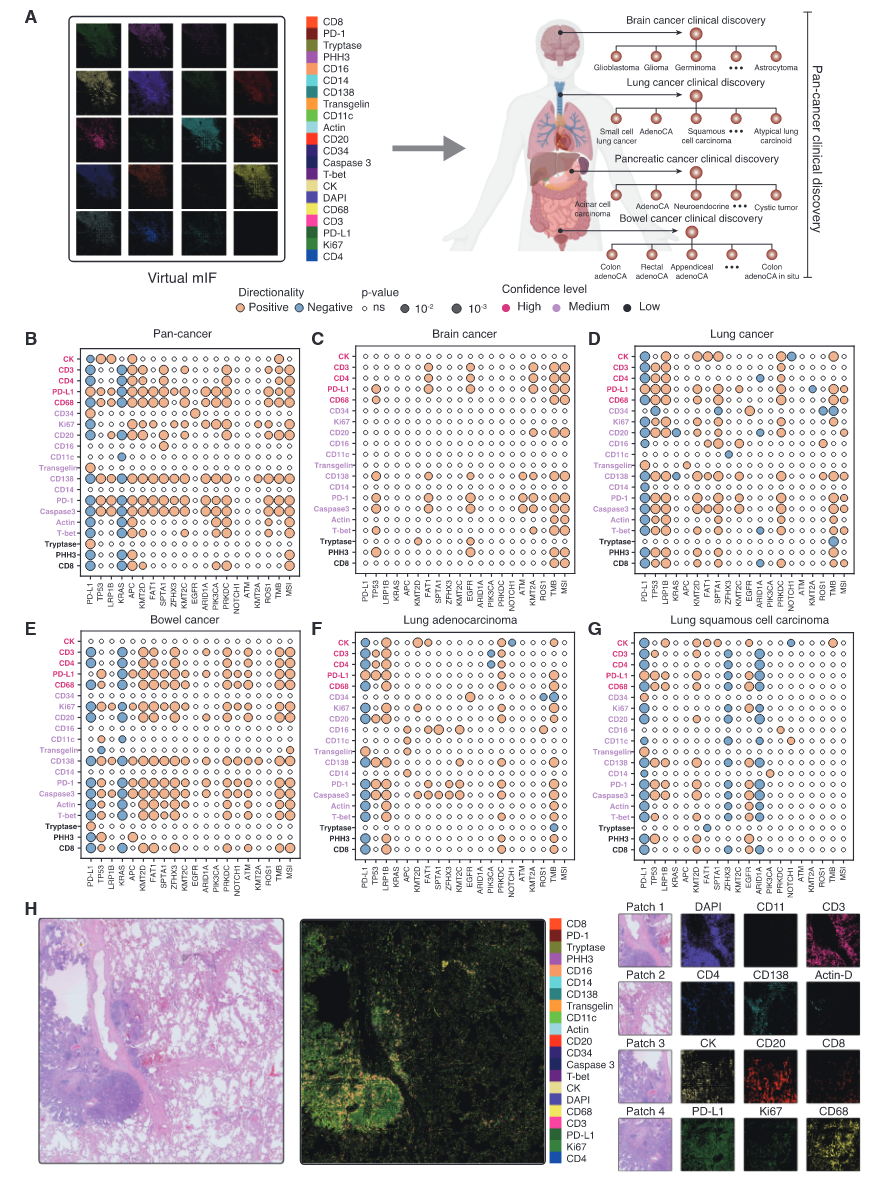
图3 GigaTIME在泛癌、癌种及癌症亚型层面识别新的TIME蛋白–生物标志物关联
虚拟人群支持病理分期与患者分层的大规模发现
在泛癌层面,GigaTIME识别出了蛋白通道与病理分期之间的显著关联(图4A)。在癌种层面,这些蛋白–分期关联在不同癌症类型之间表现出显著差异(图4B)。在肺癌中进一步开展的亚型层面分析揭示了肺腺癌(LUAD)与肺鳞状细胞癌(LUSC)之间的细微差异(图4C)。最后,为进一步评估虚拟人群的临床相关性,作者分析了虚拟mIF是否有助于根据生存结局对患者进行分层。无论是在泛癌队列(图4D),还是在特定癌种内部(图4E和图4F),GigaTIME转换得到的虚拟蛋白激活信息均能够将患者区分为具有显著不同生存轨迹的亚群。更为重要的是,将全部21个虚拟蛋白通道整合为一个综合性的GigaTIME特征,可实现更加优越的患者分层效果(图4G),凸显了不同通道之间的互补信号,并进一步验证了基于mIF的虚拟人群在临床研究中的应用价值。
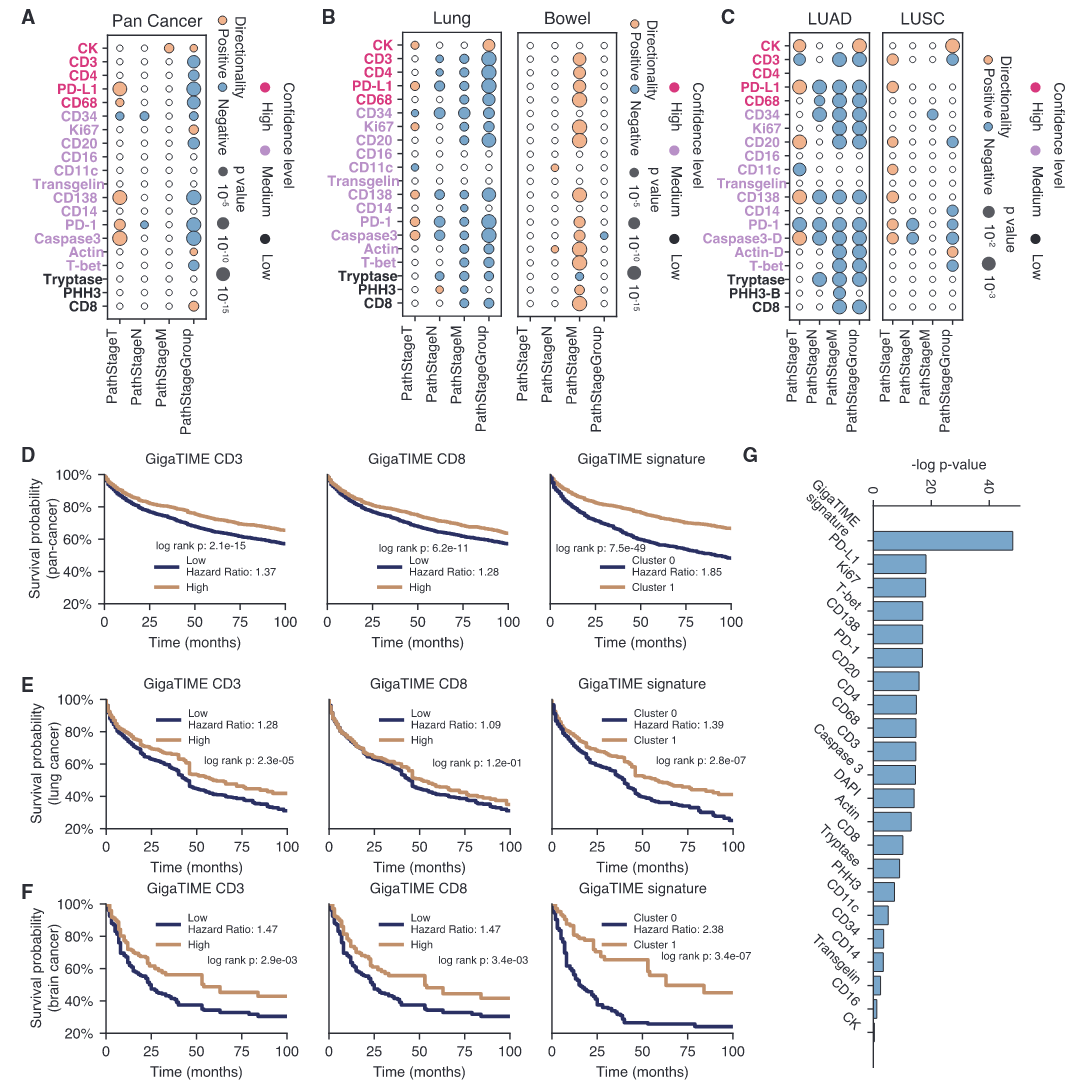
图4 GigaTIME在病理分期和生存分组中实现有效的患者分层
基于TCGA虚拟人群的独立验证
研究团队利用由TCGA构建的一个独立虚拟人群,对在Providence虚拟人群中识别到的生物标志物关联进行验证。Providence与TCGA两个虚拟人群在虚拟mIF激活水平方面具有总体一致性,其Spearman相关系数达到0.88(图5A)。此外,有80项蛋白–生物标志物关联在Providence和TCGA中均达到统计学显著,这一重叠程度具有极高的统计学意义,进一步凸显了GigaTIME的泛化能力与稳健性。此外,在癌种层面(如肺癌,图5B)以及癌症亚型层面(如LUAD,图5C),Providence虚拟人群同样揭示了显著更多的关联;相比之下,TCGA在如此细粒度层面上仅识别出极少数显著关联(图5B)。
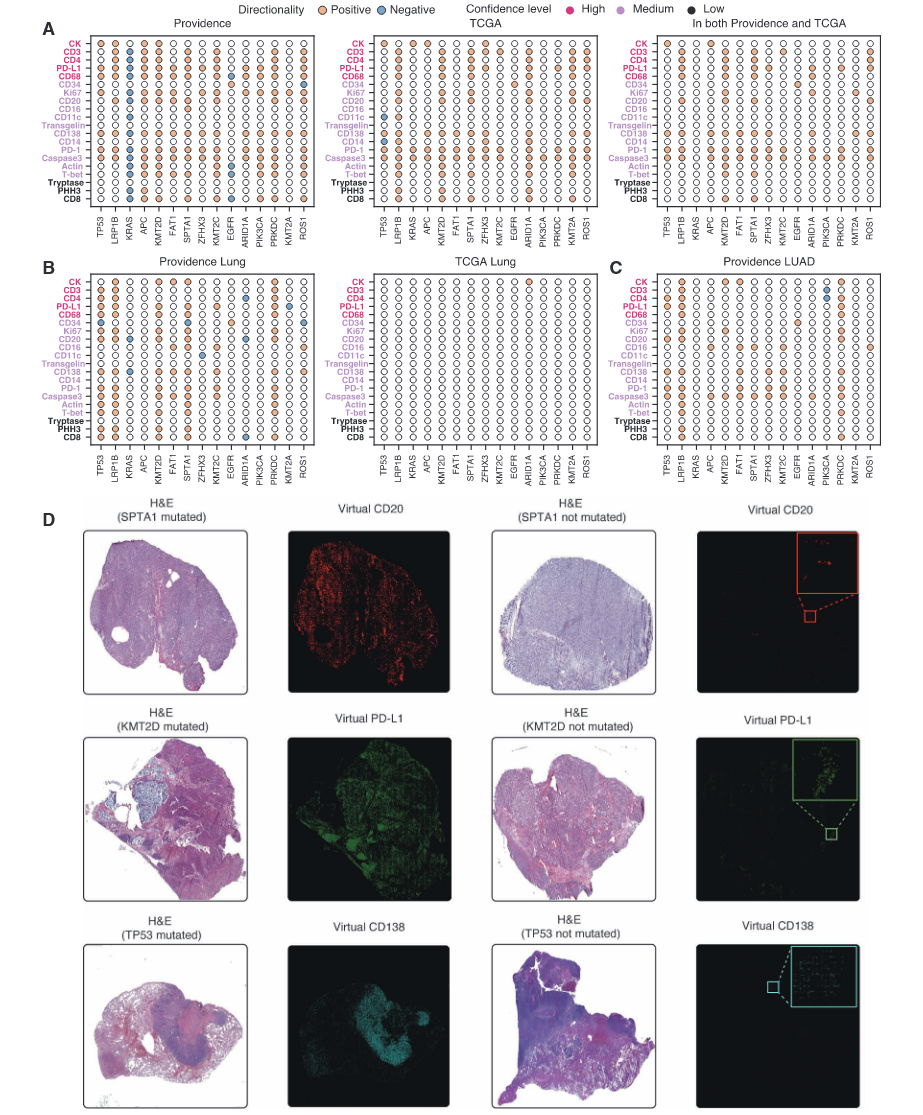
图5 基于TCGA虚拟人群的独立验证
虚拟人群揭示有趣的空间与组合蛋白激活模式
将三种标准的空间感知指标(熵、信噪比SNR和锐度)应用于Providence虚拟人群,发现它们在与特定临床生物标志物的关联中,往往比密度指标揭示更强的相关性(图6A–6C)。此外,作者使用OR逻辑运算评估虚拟蛋白的成对组合,并计算其与临床生物标志物的相关性(图6D和6E)。结果表明,组合激活相比单一虚拟蛋白能揭示更多、更强的生物标志物关联。
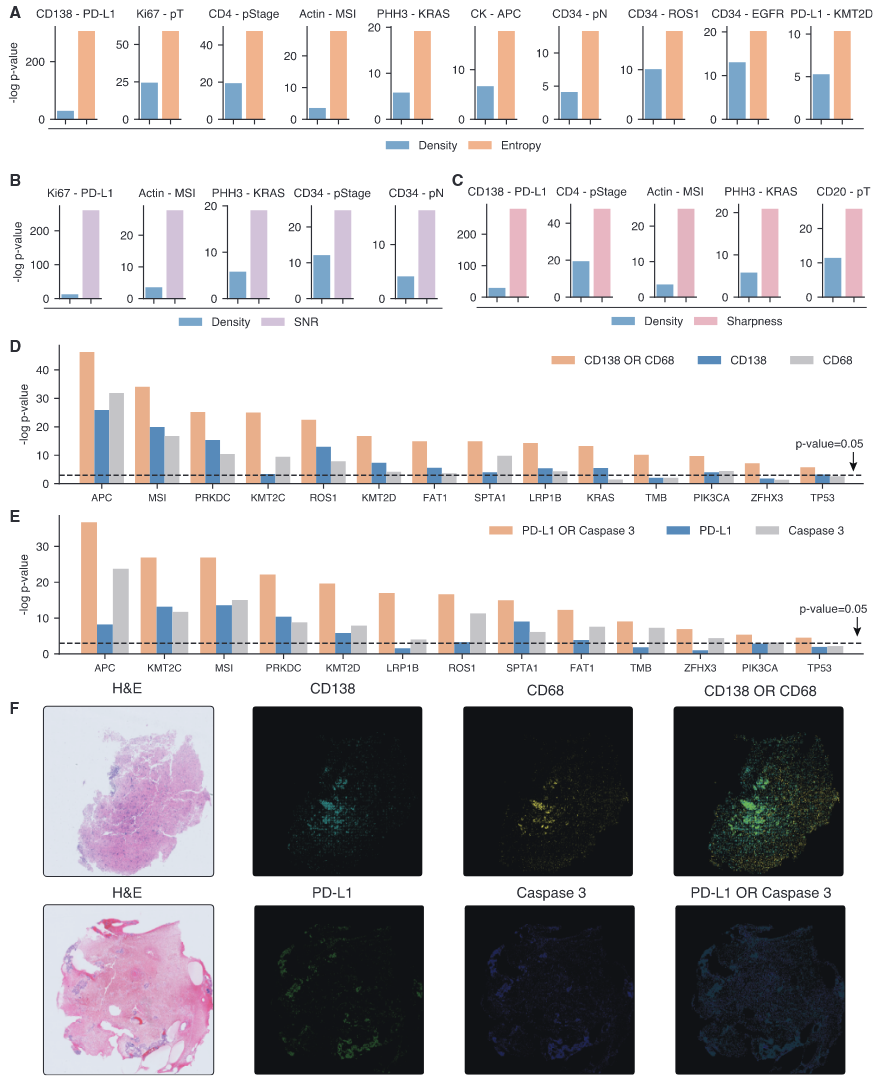
图6 GigaTIME揭示有趣的空间与组合虚拟mIF模式
局限及未来方向
本研究的关联分析基于来自51家医院和1000余家医疗机构的14256名患者,构成了迄今规模最大的虚拟mIF人群研究之一。然而,该队列中的患者主要来源于美国西部地区,地理分布和人群构成仍存在一定局限。未来仍有较大空间进一步提升患者队列在地理、种族的多样性,以更全面地覆盖当前代表性不足的人群,从而增强研究结论的普适性与临床外推价值。
值得注意的是,GigaTIME的研究结果表明,常规H&E切片中确实蕴含着丰富的、可用于空间蛋白组学建模的潜在信息。然而,并非所有蛋白信号都能被同等有效地从形态学特征中解析。部分蛋白在组织形态层面的表征并不显著,因此仅依赖H&E图像进行转换本身就存在天然上限。
研究结果已经揭示,不同蛋白通道在虚拟mIF转换质量上存在显著差异。这种差异可能源自三个因素:异质的H&E肿瘤/正常组织结构、训练数据集中阳性事件频率的差异,以及标志物特异性的技术挑战,如非特异性结合模式、表达水平差异和对组织处理方法的敏感性差异。对上述差异进行系统性量化和分析,有助于识别在转换性能上具有较大提升潜力的蛋白通道,从而为后续数据采集与实验设计提供明确方向。在未来工作中,作者计划进一步扩展可建模的蛋白通道范围,并系统评估其跨模态转换质量,逐步构建更加完整、全面的虚拟mIF图谱。
从更长远的角度看,GigaTIME的核心目标之一在于揭示肿瘤免疫微环境中细胞间复杂相互作用背后的“语法规律”。为实现这一目标,未来研究将计划把细胞分割模型进一步整合进GigaTIME的训练与推理流程中,以更深入地刻画TIME的空间组织与功能机制。
参考链接:
https://doi.org/10.1016/j.cell.2025.11.016
--------- End ---------
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢