
——研究背景——
靶向共价抑制剂(Targeted Covalent Inhibitors, TCIs)正在成为药物发现领域的一种重要模式。这类药物通过与靶蛋白中特定的亲核残基形成共价键,具有结合亲和力强、药效持久以及能靶向传统“不可成药”靶点等独特优势。然而,共价药物的理性设计仍然面临巨大挑战。一个核心难点在于如何精确预测共价蛋白-配体复合物的结构,这一过程往往涉及显著的蛋白质构象变化,传统的基于物理的计算方法(如共价对接)在处理大尺度构象重排时往往力不从心。
以AlphaFold 3为代表的生物分子共折叠(Co-folding)模型引发了结构生物学的范式转变,展示了对包括蛋白质、核酸、小分子等多种生物分子复合物的高精度预测能力。然而,这些前沿的AI共折叠模型在共价蛋白-配体复合物预测任务上的表现究竟如何?它们是否优于传统的共价对接方法?这在很大程度上仍是未知的,主要原因是缺乏一个严格、独立且无数据泄漏的基准测试集。
近日,北京大学前沿交叉学科研究院定量生物学中心/化学与分子工程学院来鲁华/裴剑锋团队在Acta Pharmacologica Sinica杂志在线发表了题为Benchmarking co-folding methods to predict the structures of covalent protein–ligand complexes的研究论文。该研究构建了专门用于评估共价复合物结构预测的综合基准测试集CoFD-Bench,并系统评估了包括AlphaFold 3在内的主流共折叠模型与传统对接方法的性能,揭示了AI模型在共价药物设计中的潜力和局限性。
——研究内容——
构建共价复合物基准数据集 CoFD-Bench
为了确保评估的客观性,避免模型训练数据的泄漏,本研究系统性地收集了2023年6月至2024年6月期间PDB数据库中发布的最新共价蛋白-配体复合物结构。经过严格的筛选和人工校验,最终构建了包含218个高质量共价复合物的基准测试集CoFD-Bench,这些结构均未被用于现有主流共折叠模型的训练。
本研究利用CoFD-Bench对三款最先进的AI共折叠模型(AlphaFold 3, Chai-1, Boltz-1x)以及三款经典的物理对接方法(AutoDock-GPU, CovDock, GNINA)进行了全面的测试。
共折叠方法 vs 经典对接方法:准确性的大幅飞跃
结果显示,共折叠方法在预测精度上显著优于传统方法。其中,AlphaFold 3表现最为出色,其L-RMSD预测精度和蛋白-配体相互作用指纹(PLIF)的恢复率均大幅领先。相比之下,传统的对接方法受限于受体构象的灵活性,即使在使用复合物晶体中蛋白部分构象进行重对接时,其成功率也远低于的共折叠方法。
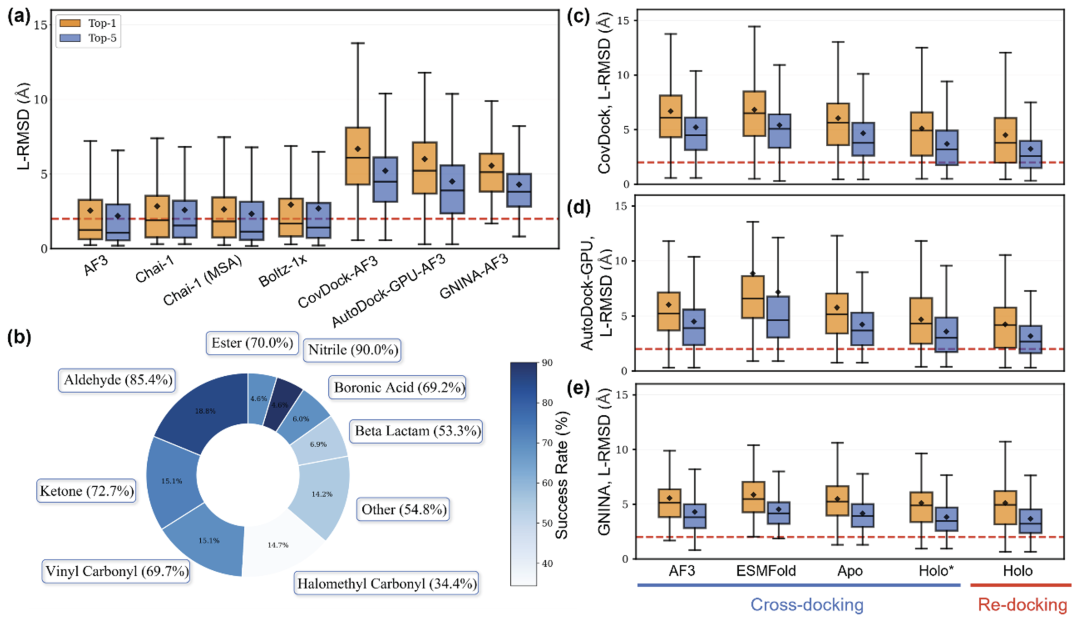
图1:(a), (c)-(e) 不同共折叠方法与经典对接方法在CoFD-Bench上的L-RMSD分布与成功率对比;(b) CoFD-Bench中配体共价头分布。
深入探究:基于“记忆”还是掌握了“规律”?
尽管共折叠方法表现优异,但在药物发现的实际场景中,模型对新颖靶点和先导化合物的泛化能力至关重要。本研究引入了SuCOS-pocket相似度指标,分析了模型性能与测试集-训练集相似度的关系。
分析揭示了一个关键局限:即使共价键的形成减少了构象的自由度,但共折叠方法的性能仍然高度依赖于测试数据与训练数据的相似性。对于那些与训练集中结构相似度较高的体系,模型预测非常准确;但面对低相似度的新颖口袋-配体对的时候模型的预测成功率显著下降,经典对接方法则不受到这个的影响。
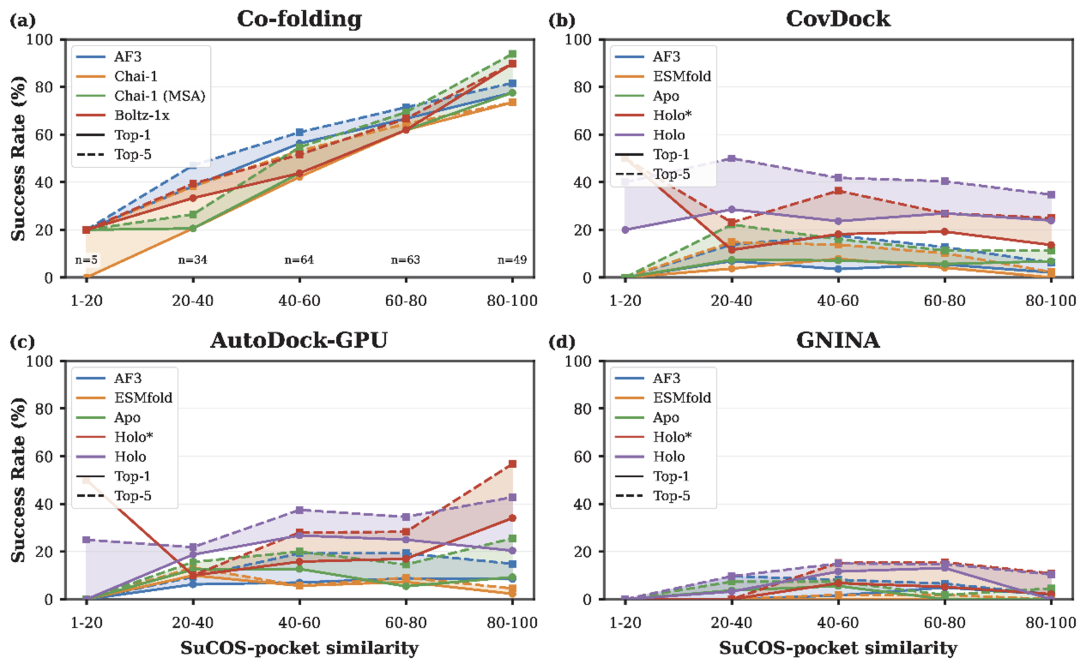
图2:共折叠方法与对接方法的成功率随训练集相似度的变化趋势。
为了展现这一点,这里展示两个具有挑战性的案例。
SARS-CoV-2 3CLpro抑制剂复合物(PDB: 8TPD): 这是一个具有新颖化学骨架的非肽类抑制剂:尽管该口袋在训练集中很常见(相似度高),但由于配体部分的新颖,AlphaFold 3未能正确预测其结合模式(L-RMSD 6.42 Å)。
VEEV nsP2蛋白酶抑制剂复合物(PDB: 8T8N): 这是一个更为独特的例子:尽管AF3在训练中分别“见过”类似的口袋和类似的配体,但这种特定的“口袋-配体组合”是全新的。结果显示,AF3完全未能重现天然结合构象,L-RMSD 10.11 Å。
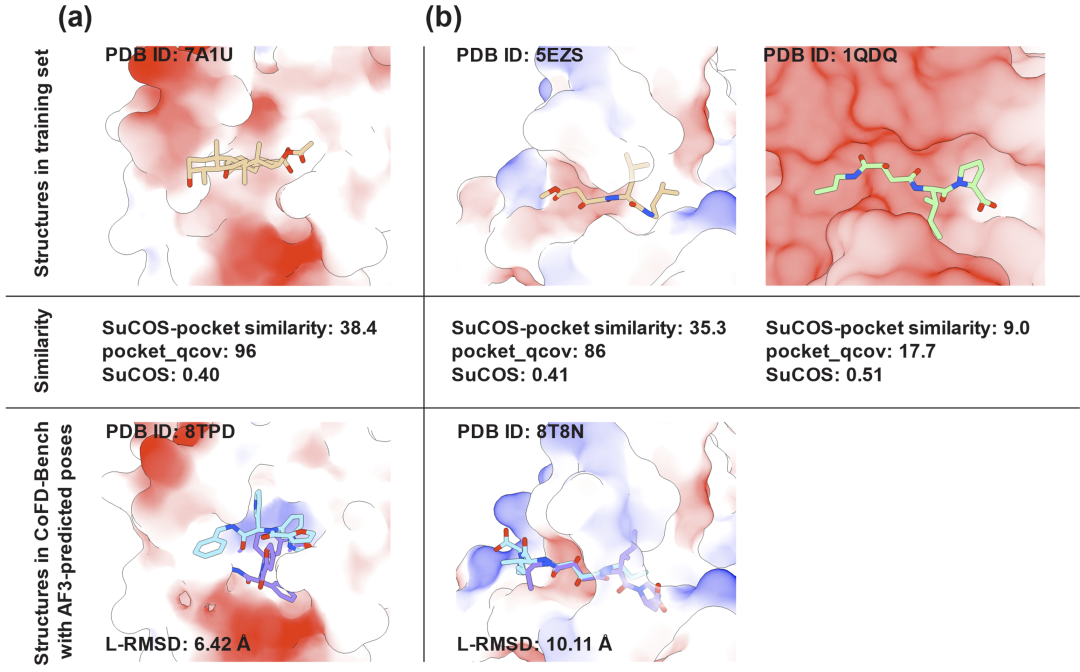
图3:两个AlphaFold 3预测失败的案例:PDB ID: 8TPD, 8T8N
上述的分析表明:当前的共折叠模型在一定程度上仍依赖于对已知训练数据/结构的“记忆”,在面对全新的化学空间时,其真实的物理泛化能力仍有待提高。
AlphaFold 3 的新能力:无需定义反应位点的“盲显”潜力
传统的共价对接需要预先明确指定反应残基,这在早期药物发现中(当反应位点未知时)是一个限制。为此,本研究设计了一个的实验:去除共价键约束,让AlphaFold 3以非共价结合的方式进行处理预测。而实验结果令人惊喜,AlphaFold 3 展现出了隐式的化学反应“直觉”,这里是两个有趣的例子:
PDB ID: 8FQU:在这个体系中,即便没有输入任何共价键连接信息,AlphaFold 3依然精准地将配体放置在了天然结合位点,并给出了近乎完美的共价结合姿态,其预测结果与显式共价预测几乎一致。
PDB ID: 7GF8:这是一个更有启发性的失败案例。虽然AF3未能将配体定位到实验确定的那个反应残基上,导致预测“失败”,但进一步研究发现,模型预测的共价弹头指向了口袋内的另一个半胱氨酸(Cys44)。
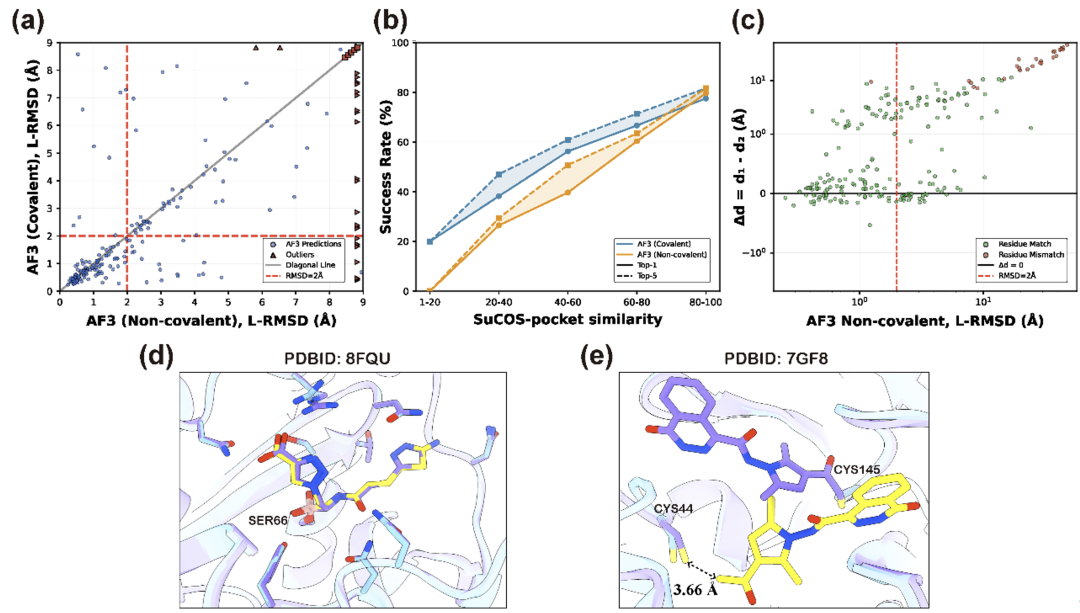
图4:基于AlphaFold 3的非共价与共价预测取得了相当的结果。(a)-(c) 两种方法在L-RMSD,成功率以及原子距离差分析;(d) AF3在无约束条件下成功复原8FQU的共价结合构象;(e) 在7GF8案例中,AF3虽然定错位点,但共价弹头指向了另一个亲核性半胱氨酸。
这意味着AlphaFold 3不仅是根据几何形状进行填充,而是可能从海量数据中隐式地学习到了亲电弹头与亲核残基之间的化学反应性模式。这一发现为未来在未知反应位点的情况下进行新的共价结合位点发掘提供了充满希望的新途径。
——总结——
本项工作建立了共价药物结构预测领域的标准测试集CoFD-Bench,填补了该领域评估标准的空白。研究结果表明,以AlphaFold 3为代表的AI共折叠方法不仅在预测精度和相互作用恢复方面显著优于传统方法,更为共价药物的设计展现了广阔的应用前景。其在不依赖先验位点信息进而识别潜在共价修饰位点的能力,将极大地拓展共价药物的靶向空间。同时,研究也客观指出了当前模型在处理新颖结构时的泛化瓶颈以及在大规模筛选中的计算效率问题(AF3单次预测耗时远高于传统对接),为未来算法的优化方向提供了重要指引。
北京大学生命科学联合中心2023级博士研究生张桐菡和定量生物学中心2021级博士研究生朱金涛为本文的共同第一作者。北京大学化学与分子工程学院来鲁华教授和北京大学定量生物学中心裴剑锋特聘研究员为本文的共同通讯作者。北京大学化学与分子工程学院博士生黄志贤和博雅博士后谢娟博士也为本研究做出了重要贡献。
该研究得到了国家自然科学基金等项目的资助。
论文信息
[1] Tonghan, Zhang et al. “Benchmarking co-folding methods to predict the structures of covalent protein–ligand complexes.” Acta Pharmacologica Sinica, https://www.nature.com/articles/s41401-025-01721-5
[2] 数据:https://doi.org/10.5281/zenodo.16466031

内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢