
真核生物基因组的三维(3D)结构通过调控元件的精准空间组织来协调细胞功能,其中染色质环作为核心结构,通过连接远端调控元件(如增强子与启动子)来调控基因表达,对维持细胞身份和正常功能至关重要;该环结构的异常断裂会导致基因表达失调,从而引发发育障碍和癌症等疾病。
目前,用于绘制染色质相互作用的实验方法如Hi-C、ChIA-PET等,虽具有开创性但成本较高、操作复杂且分辨率有限,限制了其在多物种、多条件中的广泛应用。这些瓶颈也催生了迫切需求,需开发能够从易于获取的基因组数据中准确预测染色质环的计算方法。但现有计算方法,包括早期基于特定特征的方法和近年来的深度学习方法,普遍存在物种/蛋白特异性强、模型解释性差、多模态信息整合不足等问题。因此,亟需开发一款跨物种、高精度且可解释的染色质环预测工具.
为解决上述难题,军事医学研究院伯晓晨、陈河兵团队与西北农林科技大学廖明帜团队合作提出深度学习框架CLAMP,该框架通过在大规模跨物种染色质可及性数据上进行预训练,整合了DNA序列特征、局部表观遗传信号和基因组空间距离等多模态信息。在跨10个物种、18种蛋白质和24种细胞类型的特定蛋白质介导染色质环预测任务中,CLAMP性能显著优于现有方法。同时,研究团队还开发了可解释性分析方法CoVE,可破解深度学习“黑箱”问题,揭示不同基因组特征在预测中的贡献及其生物学背景依赖性。总之,CLAMP提供了一个强大、可解释、可广泛适用的工具,能用于研究不同生物背景下的3D基因组组织,并为理解基因组功能和疾病开辟新途径。用户友好的CLAMP+ 网络平台已上线,访问地址为https://clamp.sysomics.com。

CLAMP基于Transformer架构构建,采用“预训练-微调”策略,整合了三类关键基因组特征:深度语义DNA序列表征、环锚点处的局部表观遗传信号和锚点间的基因组距离。
预训练阶段使用来自7个物种的59个染色质可及性数据集,通过掩码语言建模任务训练12层Transformer,使其学习开放染色质区域的序列语法与长程依赖关系。微调阶段融合多模态信息,引入对称多头注意力机制,整合双锚点序列特征、局部表观信号及锚点间基因组距离。最后通过参数高效微调技术(IA)适配不同任务,实现精准且灵活的染色质环预测。
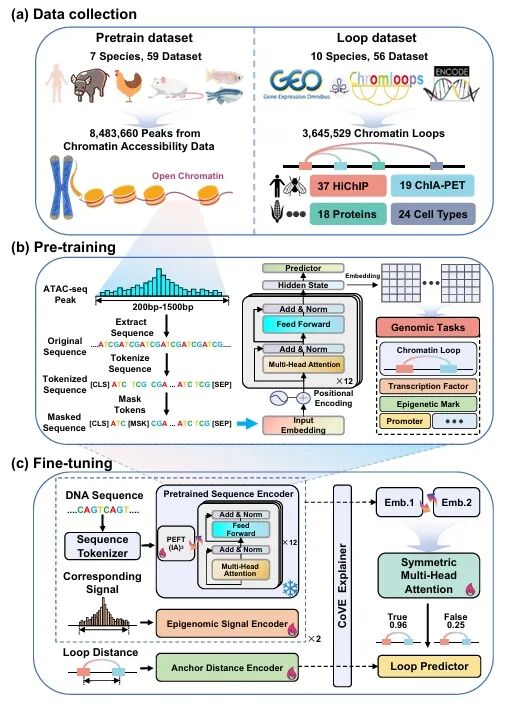
图1. CLAMP框架概述
研究团队收集了来自哺乳动物、其他脊椎动物、无脊椎动物、植物和真菌的56个染色质环数据集,以评估CLAMP适用性。结果显示,CLAMP在这些多样化数据集上均表现出高准确性。在人类细胞中,CLAMP预测不同蛋白介导的染色质环时,马修斯相关系数(MCC)均超0.9,且在不同细胞类型中性能稳定;在多物种预测中,CLAMP在哺乳动物和其他脊椎动物中AUC>0.990,在植物和无脊椎动物中分别达到0.960和0.874,即使在真菌中也优于随机预测。
进一步泛化实验表明,CLAMP能够跨蛋白质、跨细胞类型以及跨物种进行有效预测,表现出强大的适应能力,说明其学习到了超越特定因子结合偏好的结构特征。
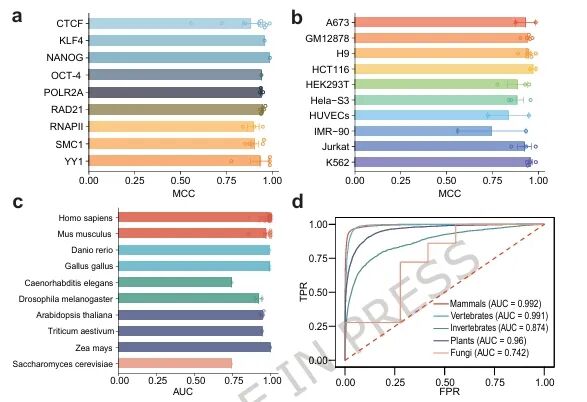
图2. CLAMP在不同生物学背景下准确预测染色质环
接下来,研究团队探究了CLAMP整合不同上下文特异性表观遗传信息以预测染色质环的能力。结果显示,通过固定模型参数并替换输入信号,CLAMP实现跨细胞类型、跨物种的精准预测,且预测结果具有明确的细胞类型特异性,证实该模型能够准确捕捉表观遗传背景的细微差异。
此外,CLAMP表现出良好的技术兼容性与稳定性。该模型可兼容不同分辨率(1-25 kb)的Hi-C数据,在低分辨率下仅出现轻微性能下降;同时支持批量 ATAC-seq、DNase-seq、ChIP-seq 及单细胞 ATAC-seq 等多种表观遗传测序数据,利用这些数据预测时MCC均超0.85。值得注意的是,即便仅使用表观遗传数据的峰区信息,CLAMP仍能高效预测染色质环子集,为低成本、便捷的染色质环推断提供了可行方案。
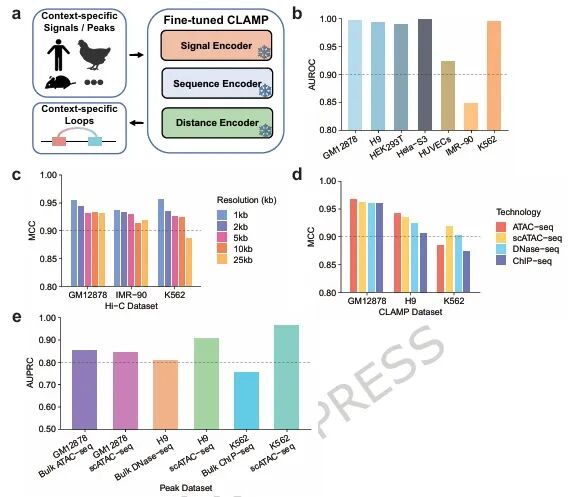
图3. CLAMP能够适应不同表观遗传环境
研究团队将CLAMP与6 种开源模型(自2021年以来发表)进行性能比较。在标准测试集Bacon上,CLAMP在多数条件下展现出最优的AUC值,即使在预测难度较高的人类甲基化修饰介导环任务中仍保持相对优势;在采用对抗性负样本构建的数据集上,其整体指标与跨物种性能均显著领先。结构鲁棒性测试揭示,CLAMP能有效适应染色质环的对称性特征,在锚点置换实验中性能保持稳定,而其他模型性能明显下降。此外,CLAMP在输入类型支持、功能兼容性(含表观整合、对称性适应、条件泛化等)及可用性上均表现突出,综合性能优于现有方法。
研究团队还在7个基因组学相关下游任务中对CLAMP进行了测试。结果显示,其性能显著优于传统CNN,与主流DNA基础模型相当,在预训练覆盖的人、小鼠物种任务中表现最优,对酵母、病毒等未覆盖物种也具较强泛化能力;CLAMP的注意力机制可跨物种精准识别CTCF、H3K4me3、RNAPII 等关键调控元件。
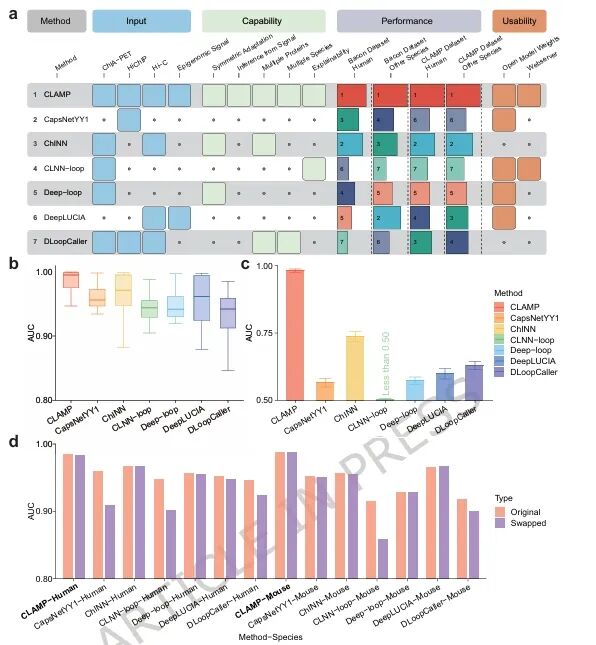
图4. CLAMP的基准测试结果
为解析CLAMP预测染色质环的决策机制,研究团队开发了可解释性方法CoVE,系统分析序列、信号和距离特征的贡献。结果显示,CLAMP的特征贡献具有显著的生物学上下文依赖性,会依据细胞类型、蛋白和物种动态调整序列、信号与距离的权重。稳定性分析表明,序列与信号特征具有较高鲁棒性,距离特征变异性较大,但其缺失导致模型性能最显著下降,证明其作为关键决定因素的重要性。
研究团队还分析了CLAMP预测结果与染色质环生物学功能之间的关联。CLAMP的高置信度预测分数与染色质环互作强度(PET计数)呈正相关,表明其能有效筛选出互作更强、潜在功能更重要的染色质环。通路分析显示,在三阴性乳腺癌细胞和果蝇胚胎细胞中,CLAMP不仅成功复现了传统方法鉴定出的核心通路,如癌症相关通路和应激响应通路,又能特异性识别出MAPK、TGF-beta等传统方法遗漏的关键通路。这表明CLAMP不仅能精准预测染色质环,还可作为传统分析的有力补充,为三维基因组调控与疾病机制研究提供更全面的功能洞见。
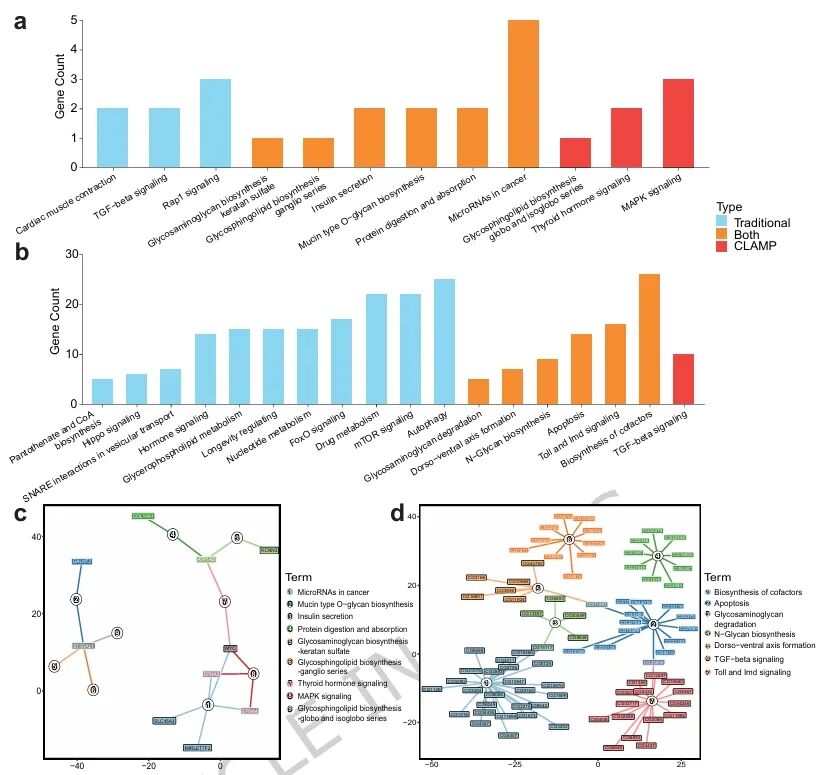
图5. CLAMP可揭示关键生物学通路
为降低使用门槛,研究团队团队开发了用户友好的网络平台CLAMP+。该平台集成两大核心模块:DNA嵌入分析模块可处理用户提交的基因组序列,生成可下载的深度特征文件并提供可视化分析;染色质环预测模块支持用户设定生物学参数、上传自定义表观数据,可生成包含统计摘要、预测列表及交互式图谱的完整报告。
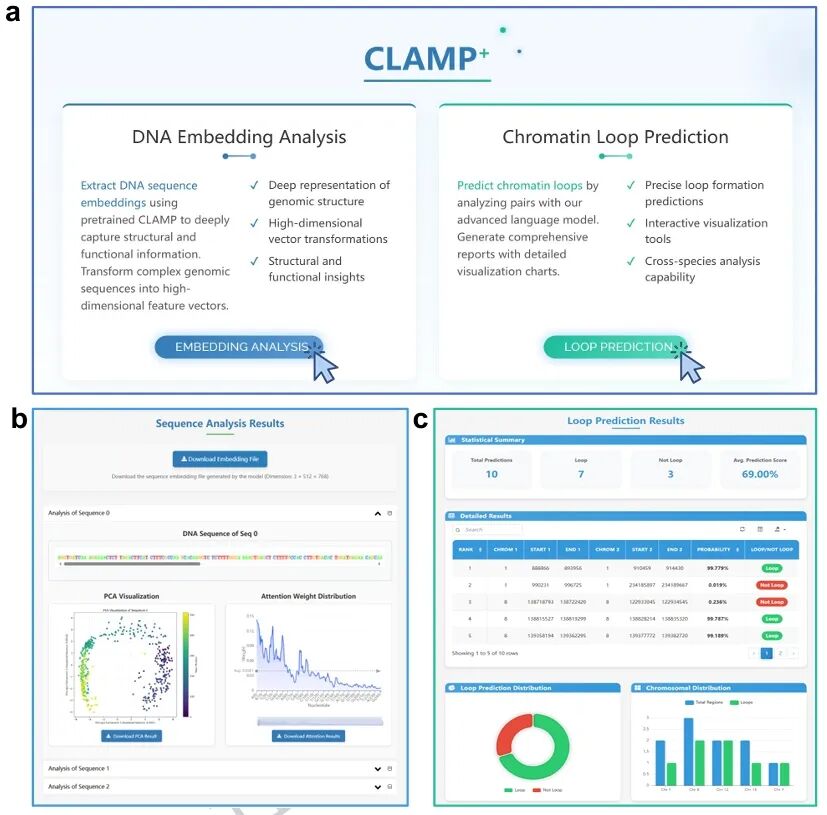
图6. CLAMP+ 网络平台
综上所述,CLAMP通过创新的语言模型架构和多模态信息整合,突破了现有方法的物种和场景限制,为3D基因组研究提供了高精度、可解释的工具;其能够跨物种、跨蛋白、跨细胞类型实现高性能预测,为进化生物学、比较基因组学及疾病机制研究提供了强大工具。CoVE解释性方法为理解模型决策与生物学机制之间的联系提供了新视角。CLAMP+平台的开放共享进一步促进了方法推广与应用落地。
参考文献:
He, Z., Sun, Y., Li, H. et al. CLAMP: predicting specific protein-mediated chromatin loops in diverse species with a chromatin accessibility language model. Genome Biol (2026). https://doi.org/10.1186/s13059-026-03948-9
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢