
研究背景
蛋白质结构并不总是“要么一样、要么完全变形”。很多真实生物学场景里,变化往往是局部、细微、但关键的。这些变化如果只看整体的全局相似度,常常会出现“整体很像,但功能位点附近其实变了”的情况;更麻烦的是,即使评分提示存在差异,研究者仍需要再花大量时间去追问:差异发生在哪?是二级结构变了,还是某个片段平移/旋转了?这类分析往往依赖人工检查与经验判断,效率和一致性都受限。
近日,浙江大学朱峰团队在Nature Communications发表MELO(Measuring and locating the changes in protein structure using MELO)。该方法瞄准结构比较的两个核心问题:变化幅度(measure)与变化位置(locate),让结构比较从“整体像不像”进一步走向“差异发生在什么位置、以何种方式发生”。

方法要点
MELO将结构变化建模为两类可解释、可定位的模式:
二级结构变化
MELO利用残基几何特征来捕捉局部构型改变,尤其适用于识别由螺旋、折叠、转角等局部结构元素发生转变带来的差异。与仅给出“是否相似”的全局指标不同,这一路径强调把变化落到残基层面的空间与序列位置:不仅能判断发生了变化,还能呈现变化在结构上的分布,从而让研究者快速聚焦到“最值得解释”的区域。
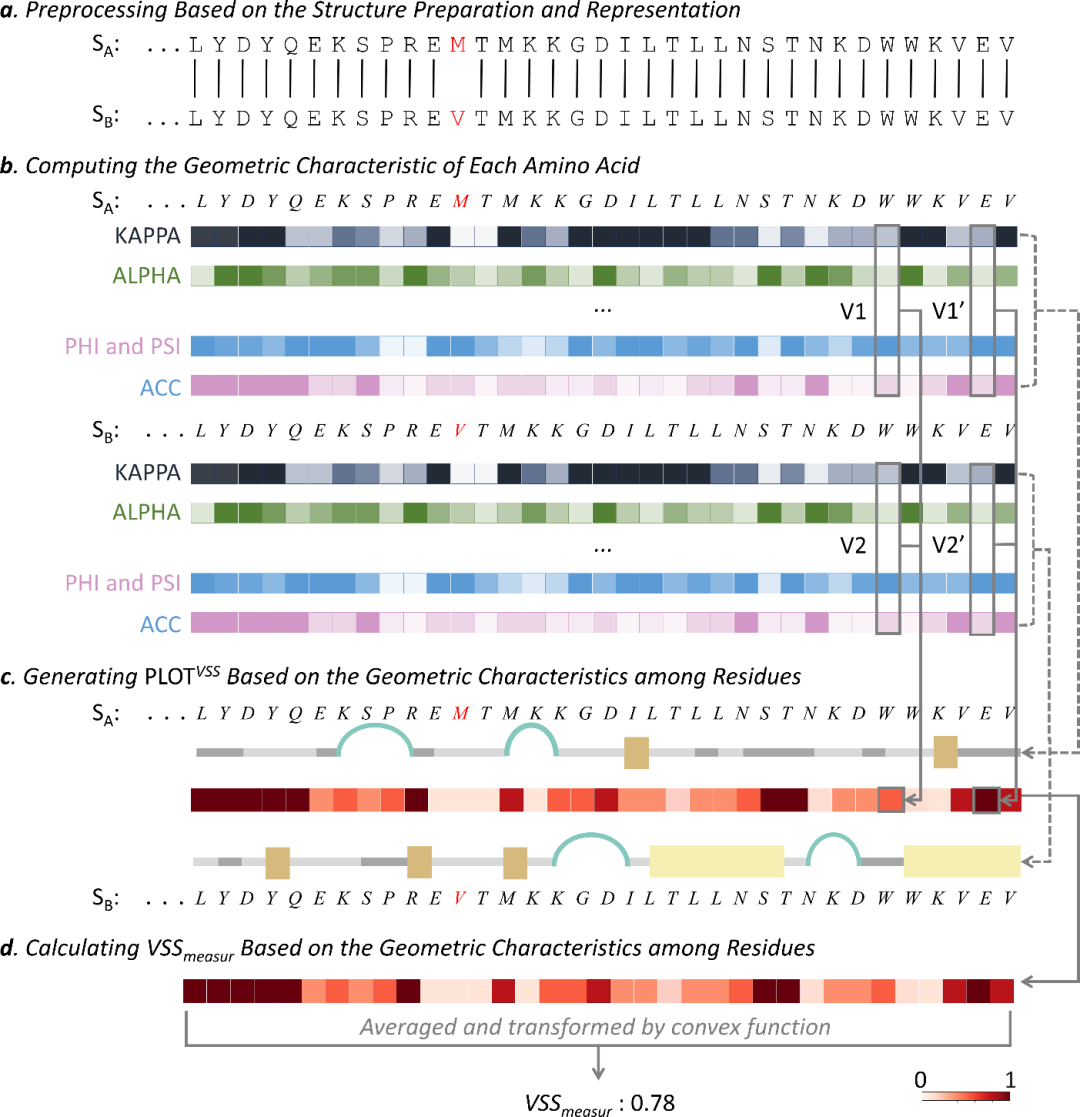
2
片段位移/重排
另一类高频变化并不一定伴随明显的二级结构类型改变,而是表现为片段级的位移、角度变化或相对位置重排。MELO通过残基相对距离变化来捕捉这类差异,能够识别诸如环区摆动、结构域相对取向变化、口袋边界移动等情况,并输出对应的定位信息。对许多机制问题而言,这类“形态没大改、位置发生偏移”的变化往往更接近真实世界的结构扰动模式。
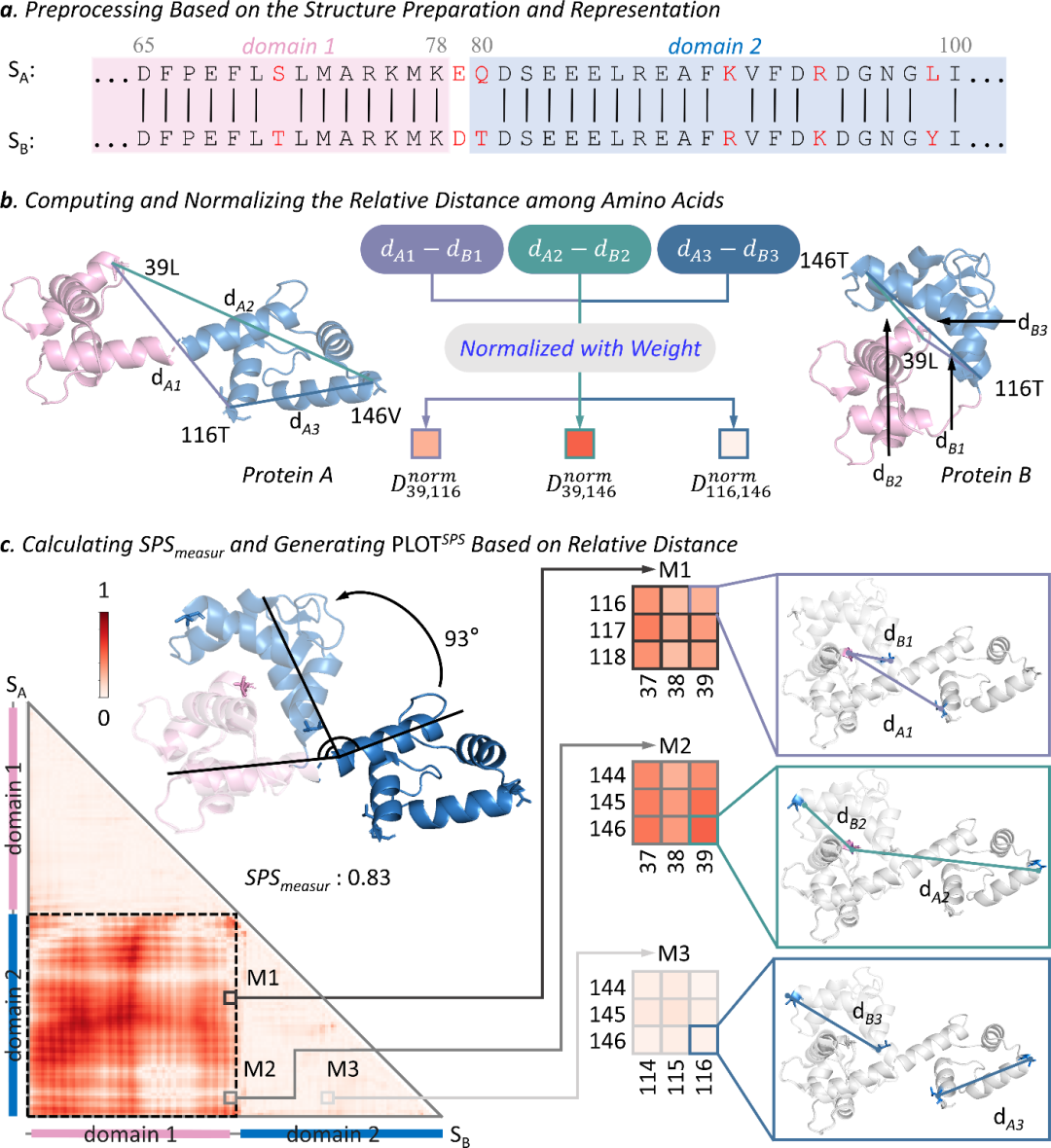
在统一判定上,研究提出MELO-score=max(两项指标),并设置阈值MELO-score≥0.5判定发生结构变化。这样的设计使得MELO既保留了两类变化的互补敏感性,又能以一个清晰的统一分数对外输出,便于在自动化筛查与下游pipeline中直接使用:例如先用MELO-score做批量过滤,再对命中的结构对进行定位与解释。
结果与对比
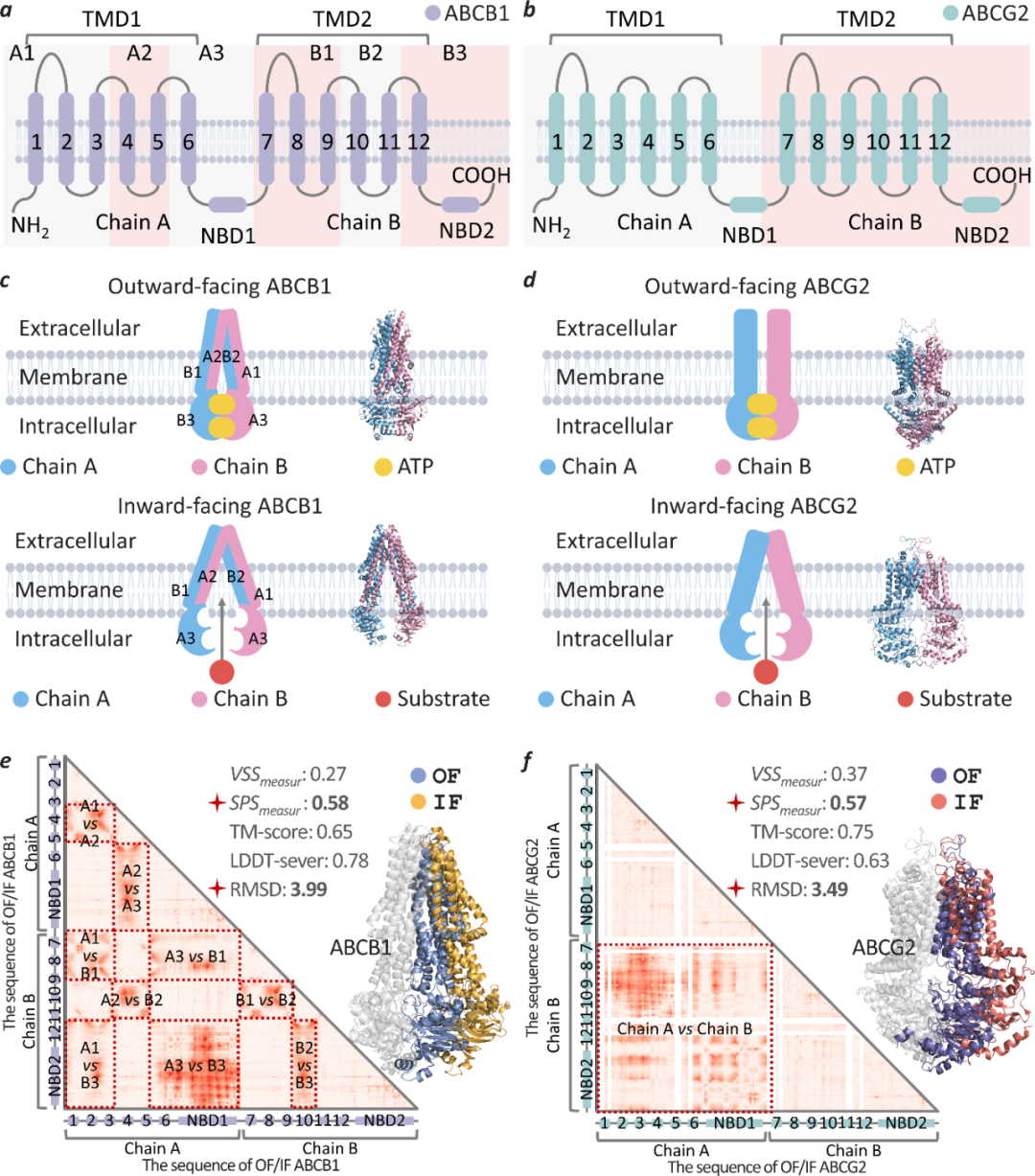
研究团队报告显示,MELO在不同幅度结构变化的捕捉上表现突出,部分场景提升超过30%。在大规模筛查中,MELO识别出 12,562 对结构变化蛋白对,而TM-score为1,411对;研究进一步强调,MELO能够识别出超过10,000个既有方法未检出的变化案例。
这些结果传递出一个重要信号:当结构差异处在“微小但关键”的区间时,仅靠传统全局指标很可能出现漏检;而MELO更倾向于把那些“全局看起来很像、但局部确实发生了结构扰动”的案例筛出来,从而让研究者有机会进一步追问其生物学意义。
更关键的是,MELO输出的不只是“有变化”的判断,还包括“哪里变了、怎么变的”的定位与类型信息。这种结果形式天然更具可解释性:研究者可以更直接地把结构差异与功能位点、相互作用界面等关注区域对齐,并据此设计更有针对性的验证实验或机制分析路径,从“发现差异”快速走向“解释差异”。
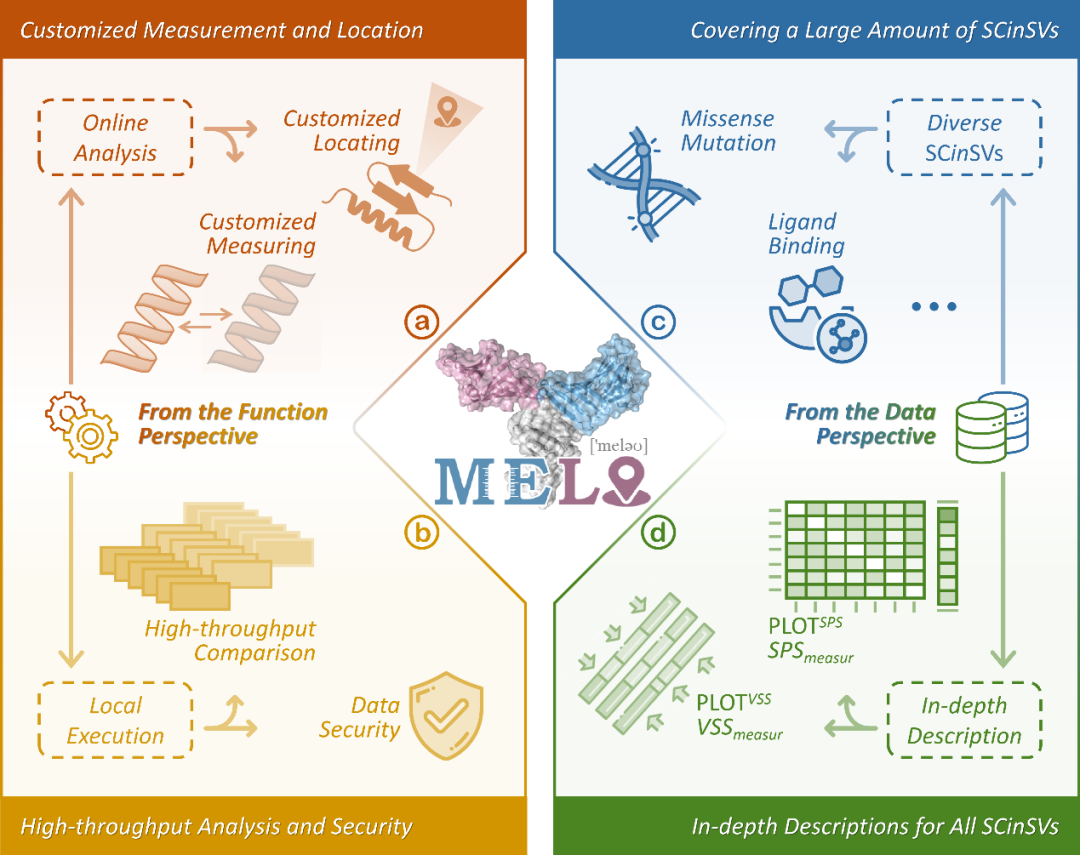
开放工具与数据
为便于社区使用与复现,团队上线MELO在线服务器并提供可下载版本:这使得不同背景的用户都能快速上手——既可以通过在线方式进行结构比较与结果查看,也可以在本地环境中开展更大规模的高通量分析或适配隐私/合规场景。
与此同时,团队还开放其识别得到的大规模结构变化数据资源。这类数据不仅能作为具体应用的候选集(例如集中分析某类扰动引发的结构变化模式),也可作为方法学研究与基准评测的基础资源,帮助后续工作在统一数据语境下进行对比与迭代,从而加速社区在“结构变化测量与定位”这一方向的持续推进。
参考资料
Zheng, L., Liao, Y., Zhang, Y. et al. Measuring and locating the changes in protein structure using MELO. Nat Commun 17, 1360 (2026).
https://doi.org/10.1038/s41467-025-68110-8
工具链接
https://idrblab.org/melo/
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢