
拷贝数变异(CNV)是基因组多样性的重要来源,也是多种疾病的重要致病因素。CNV可以直接改变基因剂量、影响调控区域,产生较大的表型效应。目前大多数全表型关联研究(PheWAS)都侧重于单核苷酸变异(SNV)和小片段插入缺失(Indel)。整合CNV可以扩大这一研究范围,为复杂疾病分析提供更深入的见解。但CNV研究仍面临诸多挑战,包括测序数据的质量问题、表型多样性以及检测罕见CNV所需的庞大样本量。
近日,阿斯利康公司研究团队通过分析470,727个英国生物样本库(UKB)全基因组测序(WGS)数据中的CNV,并结合血浆蛋白丰度、二元临床表型和数量性状数据,开展了变异与基因水平的PheWAS分析,评估了CNV在广泛人类表型和疾病中的作用。该研究是迄今规模最大的基于WGS的CNV流行病学关联研究之一,提供了有关CNV在人类疾病中作用的见解,并提供了一个丰富的高影响力基因关联资源,以促进药物靶点的发现。相关成果已发表在Nature,文章题为“Phenome-wide analysis of copy number variants in 470,727 UK Biobank genomes”。

主要研究内容
UKB拥有约50万名参与者的大规模WGS数据以及相关的健康记录数据,还包含约5万名参与者的血浆蛋白质丰度数据,能够有效克服上述CNV分析的诸多限制。研究团队使用DRAGEN v.3.7.8软件UKB WGS数据中的胚系CNV进行了分析,并将范围限定为大于10kb。最终获得的CNV数据集涵盖了来自6个不同遗传种族群体470,727个样本的80,147个重复序列和102,717 个缺失序列(图1a)。
研究进一步识别出102,717个独特的缺失片段和80,147个独特的重复片段。CNV的中位大小约为35kb;可检测的缺失序列(中位大小:26kb)比重复序列(中位大小:50kb)更小(图1c)。大多数CNV极为罕见:99.8%的变异发生率低于1%,99.3%低于0.1%。与同义突变和其他SNV相比,缺失、重复和蛋白质截短变异(PTV)在人群中的出现频率更低(图1d)。此外,大多数个体携带1- 4个检测到的罕见缺失和重复片段(图1e)。CNV长度与受影响基因的数量呈正相关,与等位基因频率呈负相关。罕见且单个的CNV往往对蛋白质编码基因产生功能缺失(LoF)效应,并且比常见CNV包含更多的剂量敏感型基因(图1f)。
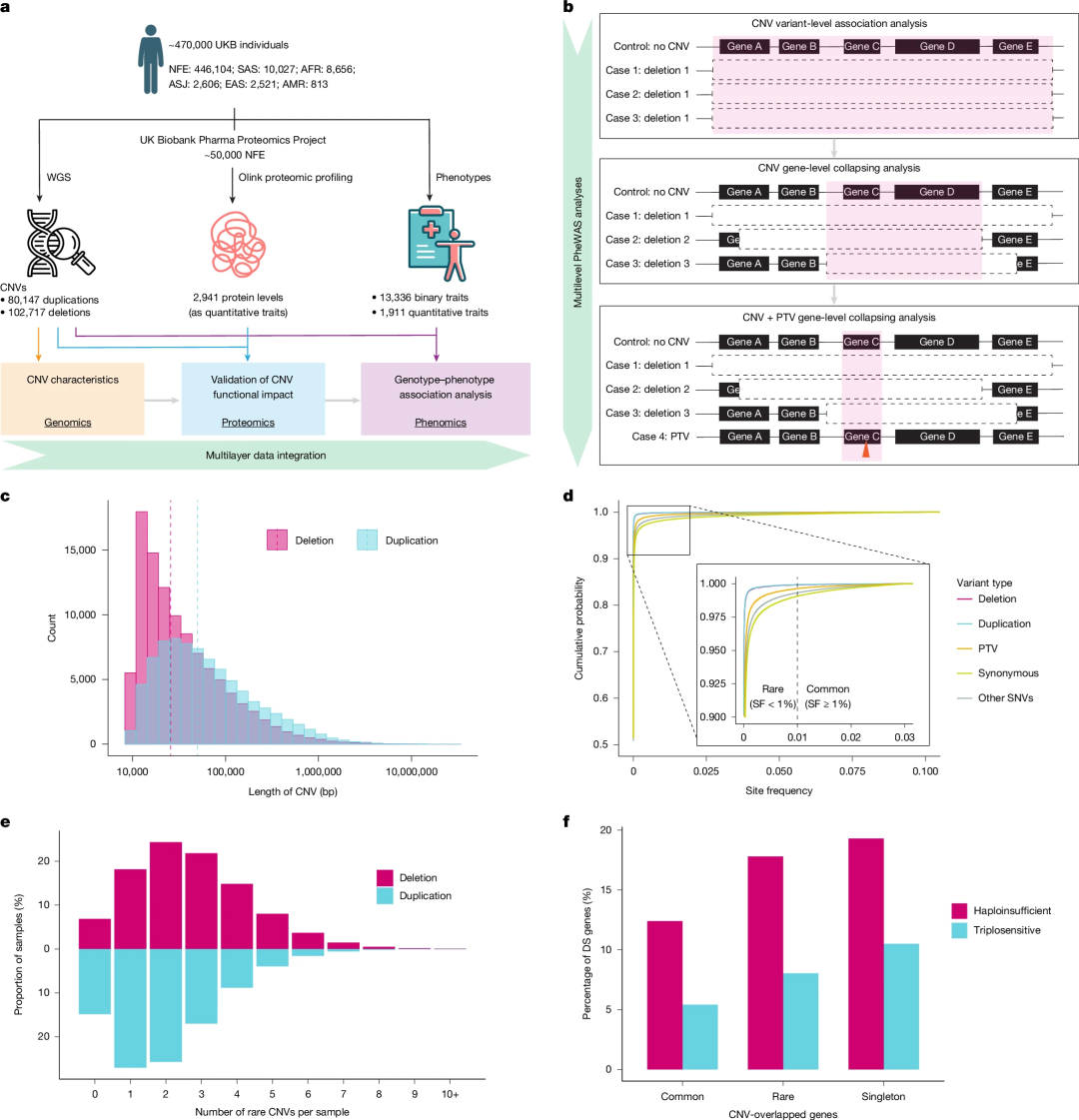
图1.470,727例UKB WGS样本的研究概况及CNV特征。
在PheWAS分析中,为评估CNV的功能影响,研究使用了三种遗传模型(显性缺失、隐性缺失和显性重复)来分析CNV与49736名个体中2941种血浆蛋白质丰度之间的关联(图2a),发现了142个显著罕见CNV -蛋白质关联(位点频率< 1%)和175个常见CNV -蛋白质关联(位点频率≥1%)。pQTL中分析发现了81个cis_1K(位于编码相关蛋白的基因1kb范围内)、20个cis_1M(1kb至1Mb范围内)和16个trans(超过1Mb范围)罕见CNV pQTL。cis_1K类型CNV pQTL的功能效应与剂量效应一致:47个罕见cis_1K缺失中有46个(98%)导致蛋白水平下降,34个罕见cis_1K重复中有31个(91%)导致蛋白水平上升(图2b)。常见CNV pQTL对蛋白质丰度的影响与罕见pQTL相似,但平均效应规模较小;罕见CNV具有更大的影响。
研究将已报道的显著小变异pQTL与该分析的CNV pQTL进行了比较。罕见CNV比常见CNV具有更大的效应(图2d)。值得注意的是,研究发现了二元性状与定量性状之间的交叉验证关联:有14个CNV在二元表型和定量性状中均具有显著性(图2g)。此外,有3个非编码CNV显著与4种二元表型相关,还有39个与126种定量性状相关。
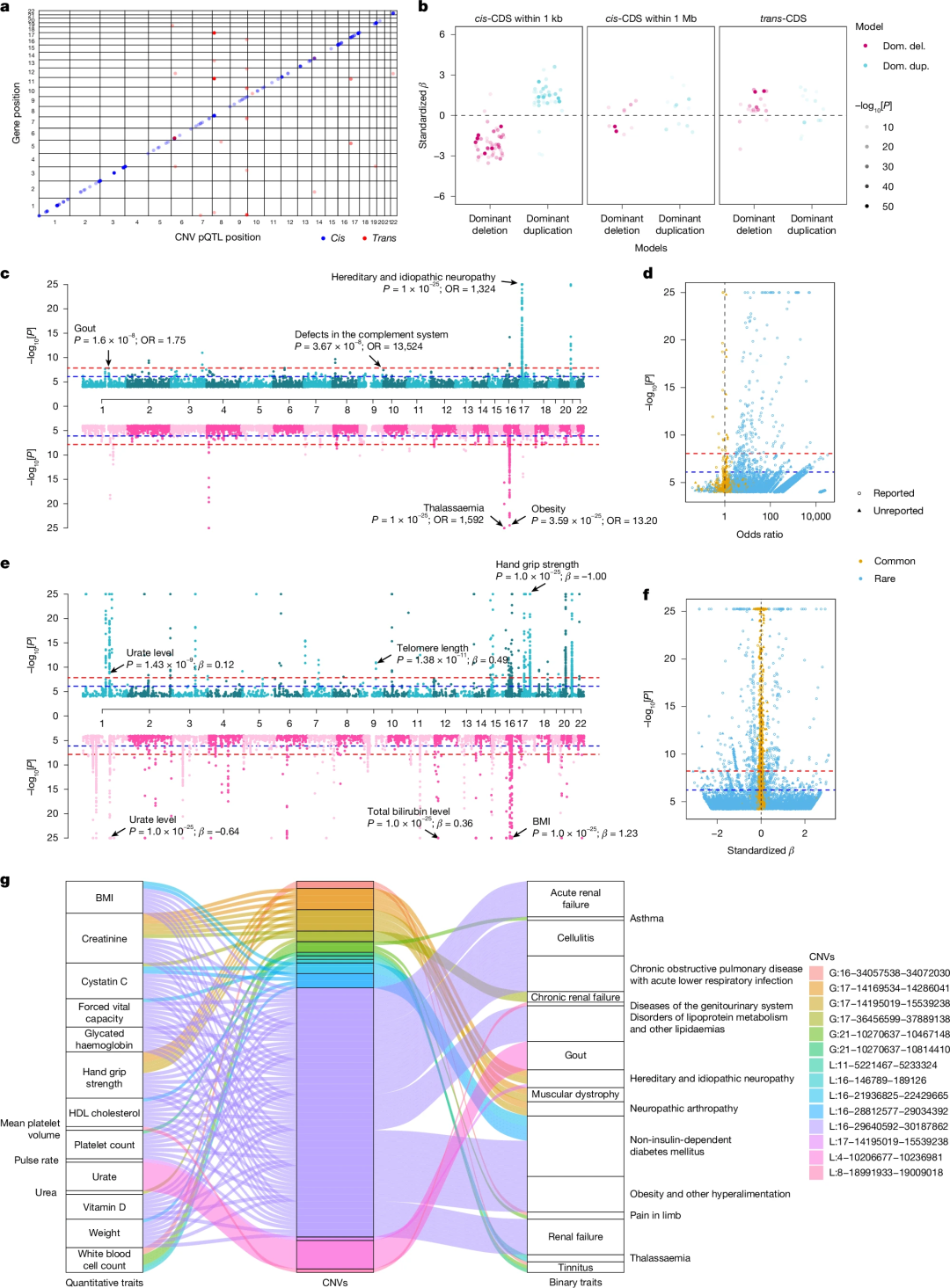
图2.CNV水平的PheWAS分析
随后,研究团队进行了CNV基因水平的整合分析,以确定与特定表型相关的基因关联。分析确定了166个显著的基因-蛋白质关联,包括101个(61%)cis-CDS关联和65个(39%)trans-CDS关联。cis-CDS的影响符合预期(图 3a):整个基因拷贝数的增加会提高蛋白质水平,而任何编码部分的缺失则会降低蛋白质水平,其中某些位点在缺失和重复情况下的效应呈现出相反的特征(图3b)。基因层面的整合分析又发现了17个新的顺式编码序列pQTL。
研究团队对470,727个基因组中的13,336个二元性状进行了基因水平的CNV整合分析。结果显示,共有17个基因簇在177个二元性状中贡献了196个显著关联(图3c),其中11个簇包含2个或更多的基因。最大的簇包含位于16号染色体上的40个基因,与多种疾病相关,包括2型糖尿病、贫血、肾衰竭、精神和行为障碍、呼吸系统疾病。此外,分析还发现了8个新的位点,涉及102个二元表型关联,这些关联在变异层面均不具有统计学意义(图 3d)。研究发现了617个显著关联,涉及77个基因簇和287个定量特征(图3f);在变异水平的分析中,有342个定量关联未被检测到(图3g)。
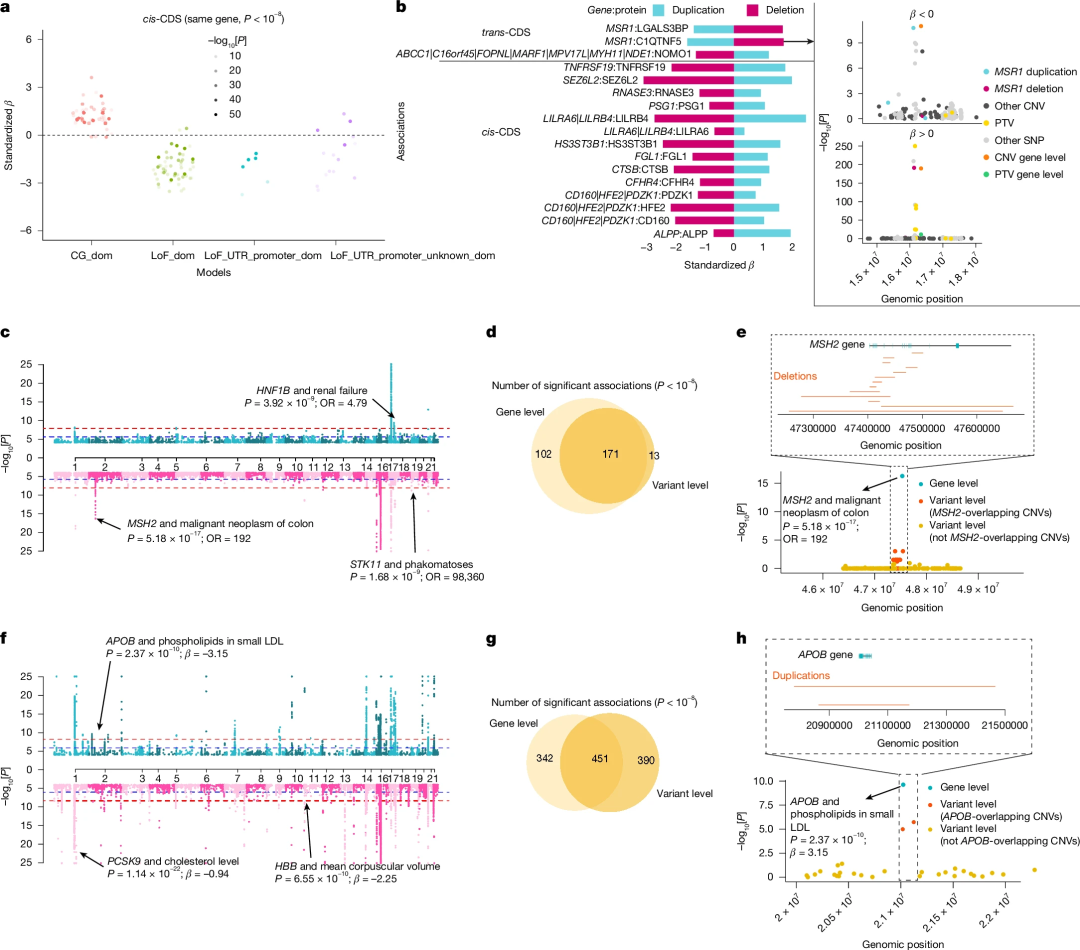
图3.CNV基因水平整合分析。
该研究显示,大多数CNV缺失所产生的致病变异效果与PTV类似。因此,研究人员引入了一种CNV+PTV基因层面的合并模型,将CNV缺失与每个基因中符合条件的PTV进行整合,然后分析合并效应与Olink蛋白水平之间的关联。该模型检测出了39个显著的cis-CDS pQTL和34个显著的trans-CDS pQTL,这些是单独的PTV和CNV模型所遗漏的。进一步分析表明,该综合模型显示出更高的效力和更清晰地阐明致病基因的能力,同时,这一增强的基因水平检测为显著提高pQTL发现潜力提供了机会,并在相同数据集中扩展了以前未知的反式关联。
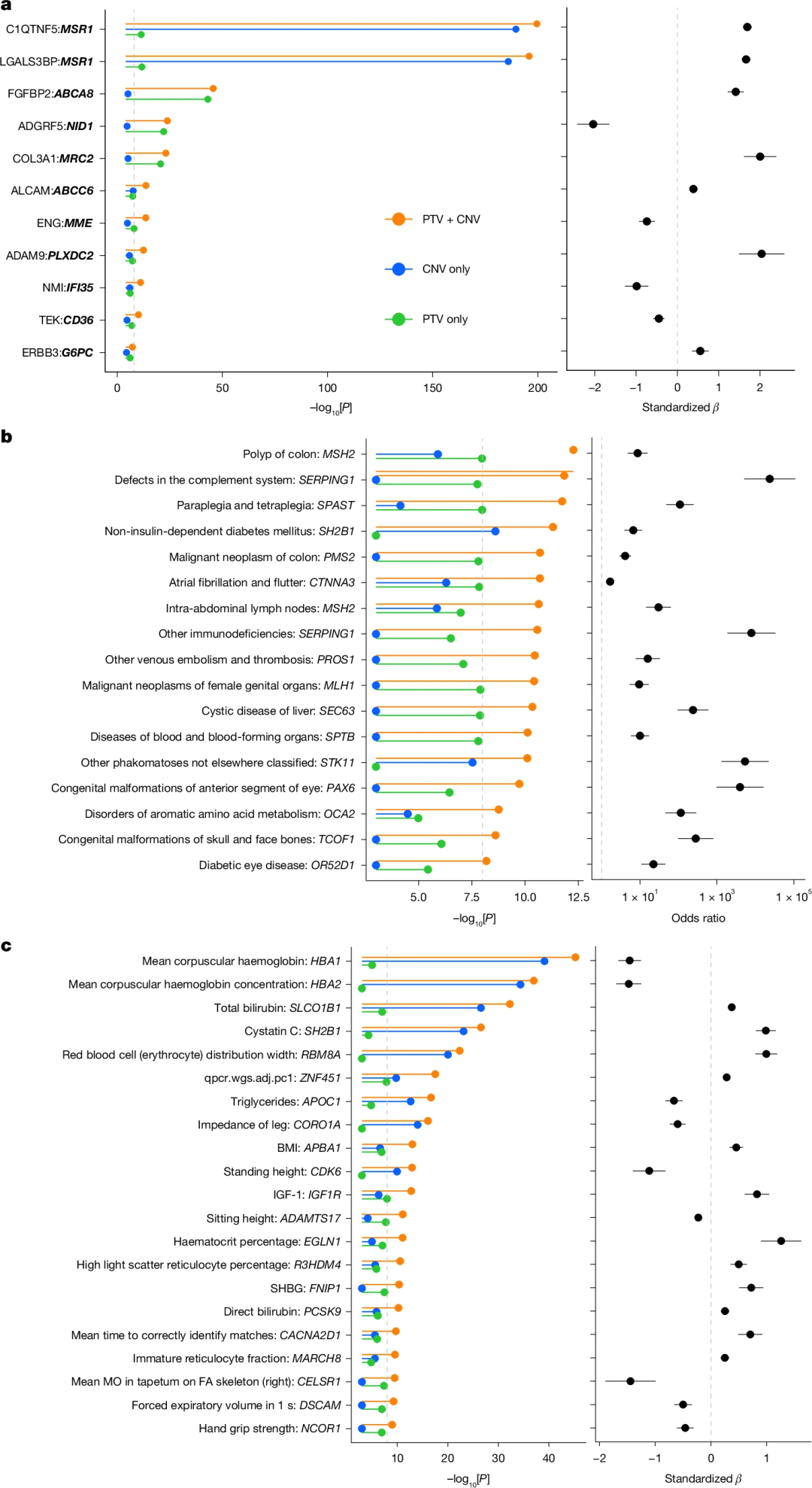
图4.PTV+CNV基因水平整合分析。
最后,研究人员针对二元和定量性状,进行了基于种族的CNV阵列关联性研究和荟萃分析,同时在变异和基因层面进行了此类分析,从而实现了族群特异性的发现。
结 语
UKB大规模的基因组、蛋白质组和深度表型数据的集合,使得能够以前所未有的规模进行CNV对健康和疾病影响的系统和多组学评估。这一基于WGS的CNV PheWAS分析将基因组学、蛋白质组学和表型组学与变异水平、CNV基因水平以及CNV+PTV基因水平模型相结合,确定了与疾病相关CNV直接关联的蛋白质,并展示了CNV如何影响人类健康和疾病。
该研究涵盖了人类多种二元和定量表型,能够识别已知和此前未知的与疾病相关的区域,包括疾病风险和保护区域,发现SLC2A9基因增强子非编码区缺失与痛风风险降低相关。值得注意的是,研究纳入了近期可用的端粒长度相关表型,发现了ZNF451基因中与白细胞端粒长度增加相关的1个罕见缺失。该数据资源将为进一步功能和机制研究提供宝贵的参考依据,为人类疾病生物学提供见解,并扩大新药物靶点和生物标志物的发现范围。
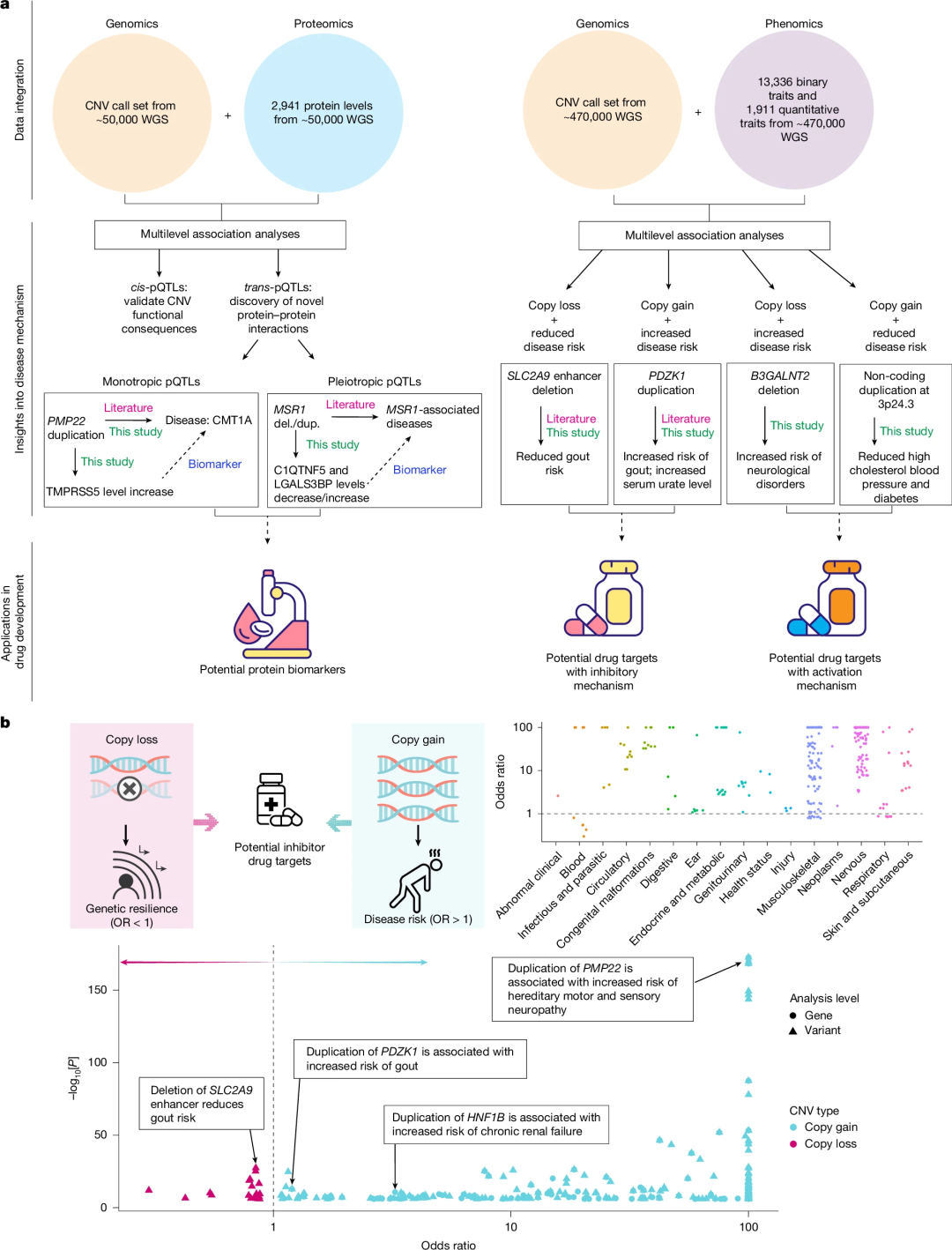
图5.CNV PheWAS有助于发现新的药物靶点和生物标志物。
原文信息:
Zou, X.Z., Hu, F., Lou, H. et al. Phenome-wide analysis of copy number variants in 470,727 UK Biobank genomes. Nature (2026).
https://doi.org/10.1038/s41586-025-10087-x
·END·
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢