

作者:W. Patrick Walters等(原载《J. Med. Chem.》2011年)
尽管药物研发成本急剧上升,但以每年获批新药数量衡量的生产力在过去十年间一直停滞不前。人们提出了许多不同的解释来说明这种下降,例如监管审查日益严格、超大型组织固有的低效性、对基因组学识别出的高度投机性靶点的追求,以及对将边缘化合物推进到后期临床试验做出更客观的决策。
常被引用来帮助解释药物生产力低下的另一个因素是,矛盾的是,药物化学家合成的许多化合物并不“类药”,至少根据“类药五原则”或REOS等各种标准来衡量是如此。这一发现可以从许多不同角度来合理化,如下所述。
为了更好地理解哪些因素对药物化学实践影响最大,我们推测,研究药物化学家所合成分子特性随时间的变化可能具有启发性。为此,我们分析了《JMC》第1至52卷(1959–2009年)间报道的分子。本次评估共包含了415,284个分子。
自1959年以来,《JMC》一直是学术界和工业界药物化学研究组报告的主要来源。它是药物化学领域被引用最多的期刊,2009年记录的被引用次数超过42,000次。虽然已有多个研究组对上市药物或近期药物专利中的化合物进行了分析,但我们尚未见到对大量药物化学文献进行类似分析的报告。对药物的分析未必能代表那些未能进入市场的、数量更为庞大的药物化学化合物。药物化学合成化合物与获批药物的比例估计值差异很大。大型制药公司的药物化学研究组每年合成超过100,000个化合物;然而这些公司每年通常仅能获得少数几个新药的批准。其他机构可能声称其生产力更高,但我们可以有把握地假设,每个获批药物对应的药物化学合成化合物数量远超10,000个。显然,并非药物发现计划中合成的每个化合物都会成为出版物的一部分,但我们可以认为,与每年获批或进入临床试验的少量药物相比,JMC上的出版物能提供对该领域更广泛的概览。
为了近似估算JMC文章所代表的药物化学产出的比例,我们计算了JMC化合物与Vertex Pharmaceuticals公司在1990年至2010年间药物化学项目产出的相似性。大约5%的Vertex化合物与至少一个JMC化合物具有至少0.7的Tanimoto相似性(Daylight指纹)。对于更大的机构,这个比例可能更高。尽管JMC的覆盖范围并不全面,但我们认为它足以代表药物化学的整体趋势。
过去10年中,多个研究组对上市药物和临床研究化合物的特性进行了分析。Lajiness在2004年的一篇综述中对这项工作进行了全面总结。Vieth 2004年的一篇论文显示,1982年至2002年间上市药物的分子量中位数和CLogP变化极小。然而,当按时间顺序对药物进行分组并考虑不同时间段时,可能会得出不同的结论。2004年,Leeson和Davis发表了一项分析,比较了1983年之前上市的口服药物(864种)与1984年至2004年间上市的口服药物(329种)之间的差异。他们发现平均分子量、氧原子和氮原子数量、环数量以及可旋转键数量均有所增加。然而,1984年前后的药物在CLogP、极性表面积百分比和氢键供体数量方面似乎没有统计学上的显著差异。
Leeson及其合作者的两篇论文将主要制药公司近期专利中化合物的计算特性与一组上市口服药物的特性进行了比较。在2007年的论文中,作者发现随着时间的推移,药物中的分子量和氢键基团数量显著增加,而药物CLogP的增加则较为温和。作者还发现,近期药物专利中的化合物比1990年以来批准的药物更大(中位分子量451 vs 432)且更具亲脂性(中位CLogP 4.1 vs 3.1)。在2010年发表的文章中,作者研究了计算特性与离子类别随时间变化的关系。他们发现,除1990年后批准的酸性药物外,药物的CLogP中位数、极性表面积和氢键供体数量自1960年代以来一直相对稳定。虽然药物中的氢键受体数量有所增加,但在形状或手性方面没有随时间变化的趋势。作者再次发现,当前药物专利中的化合物往往比口服药物更大、更具亲脂性且三维性更差。对2000–2009年间药物专利中出现的化合物的考察显示,分子量、CLogP和形状没有随时间变化的趋势,作者提出这些特性的膨胀可能正在趋于平缓。
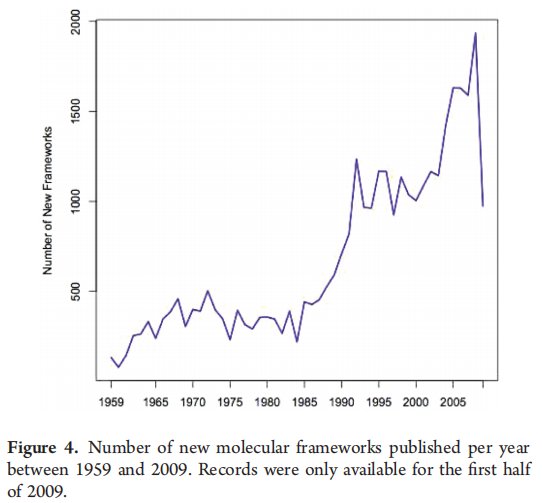
本研究使用的数据通过合并两个数据库的数据集获得:MedChem数据库(GVK Biosciences,印度海得拉巴)和EMBL的公开ChEMBL数据库。从MedChem数据库中提取了1959年至2007年间发表在《药物化学杂志》上的18,629篇文章中的408,330个化合物的化学结构和发表年份,保存为SD文件。从ChEMBL数据库第2版中提取了1980年至2009年间发表在14,447篇JMC论文中的335,640个化合物的化学结构,保存为SD文件。使用QuacPac工具包(OpenEye Scientific Software, Santa Fe, NM)为每个分子的标准互变异构体生成SMILES字符串,并用于创建422,678条记录,对应于SMILES、卷号和起始页面的唯一组合。排除了7645个分子量大于1000的化学结构,最终剩下来自19,299篇论文的共415,284个结构用于本次分析。对JMC的415K结构集与2009版MDL药物数据报告(MDDR)(Symyx, San Leandro, CA)重叠部分的计算表明,本分析所用的化合物包含了512种已上市药物和258种曾进入临床试验的化合物。数据库中每年结构的数量如图1所示。请注意,在本分析使用的ChEMBL数据库版本中,2009年的记录仅包含上半年数据。每篇文章结构数量的汇总统计见表1。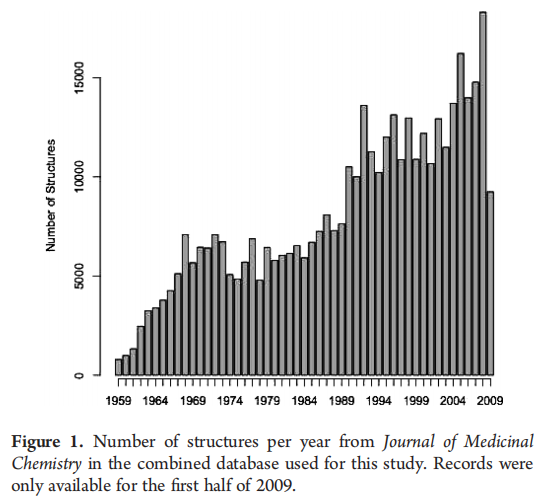

为了进行比较,从2009版MDDR数据库中提取了一组1219种分子量小于1000的上市药物。选择了所有“PHASE”字段设置为“Launched”的条目。药物的批准年份取自“PHASE_YEAR”字段中最早的条目。为这组药物计算了与JMC集相同的特性。这些特性在图2和图3中以虚线表示,均值周围的95%置信区间以灰色区域绘制。
分子量、氢键基团数量和可旋转键的计算是使用内部开发的Python脚本进行的,这些脚本利用了OEChem工具包(OpenEye Scientific Software, Santa Fe, NM)。表2中的SMARTS模式用于识别这些基团。
极性表面积的计算使用了一个内部开发的C++程序,该程序采用了Ertl最初发表的方法。该程序也使用OEChem工具包处理所有分子操作功能。log P计算(CLogP)使用内部开发并验证的XLogP算法实现来完成。分子复杂性使用MDL 166位指纹进行评估,该指纹通过OpenEye Scientific Software的OEGraphSim工具包实现。
所有统计分析和图表均使用R语言(版本2.11.1)生成。图2和图3中的图表以实线显示了在特定5年区间内发表的论文中出现的化合物的平均值和中位数。平均值用彩色方框表示,蓝色方框表示与前一区间存在统计学显著差异(均值差异的p值≤0.05),红色方框表示缺乏统计学显著性(均值差异的p值>0.05)。统计学显著性的计算使用R函数“pairwise.t.test”进行。该函数执行多重成对比较,并使用Holm方法来校正多重检验。
分子量与药物发现中的许多关键参数相关。据报道,过去几十年中,上市药物以及候选药物的分子量一直在增加。Lipinski表明,在1962年至1997年间,默克和辉瑞提出的临床候选药物的分子量显著增加。Leeson和Davis显示,1983年至2002年间批准的口服上市药物的分子量比1983年前批准的药物高出14%。在2008年的一项研究中,Gleeson使用主成分分析法确定了一组最优描述符,用于对葛兰素史克常规进行的12项ADME试验建立预测模型。分子量是所测试描述符中最显著的,解释了第一个主成分中92%的变异。这些报告,加上早期临床试验的高失败率,使得人们重新关注物理性质,特别强调更低的分子量化合物。这种兴趣的复苏似乎部分是由Lipinski发表的“类药五原则”所推动的,该原则帮助定义了已成为许多药物发现努力中常见部分的性质范围。人们对类先导性也重新产生了兴趣,这推动了近期基于片段的设计以及配体效率和“三原则”等策略的发展。
分子量与选择性之间的关系仍存疑问。辉瑞最近的一项研究表明,较高分子量的分子往往更具选择性,而诺华的一项类似研究则得出了相反的结论,即较大的分子往往更具混杂性。今年发表的第三项研究则表明,对于百时美施贵宝合成的激酶抑制剂,分子量并非选择性的决定因素。靶标和药理学的差异可能使得很难对像选择性这样复杂的事物进行概括。
图2显示了1959年至2009年间JMC论文中出现的化合物的分子量趋势,按5年间隔划分。JMC化合物的趋势与药物的趋势相似。对于JMC化合物,平均值和中位数均随时间出现统计学上的显著增加。在1960年代,JMC化合物的中位分子量接近300(1959–1964年和1965–1969年均为303)。在1970年代,中位分子量每5年增长2–3%,上升到1970–1974年的317和1975–1979年的326。分子量的增加在整个1980年代以相似的速度持续。分子量最显著的增加发生在1990年至1994年期间,中位分子量比前一个5年期间增加了10%。在1995年至2009年期间,分子量的增加较为温和,中间5年期间的增长在0–2%之间,最近一个时期(2005–2009年)的中位值为388。总体而言,在整整50年间,JMC化合物的平均分子量增加了25%。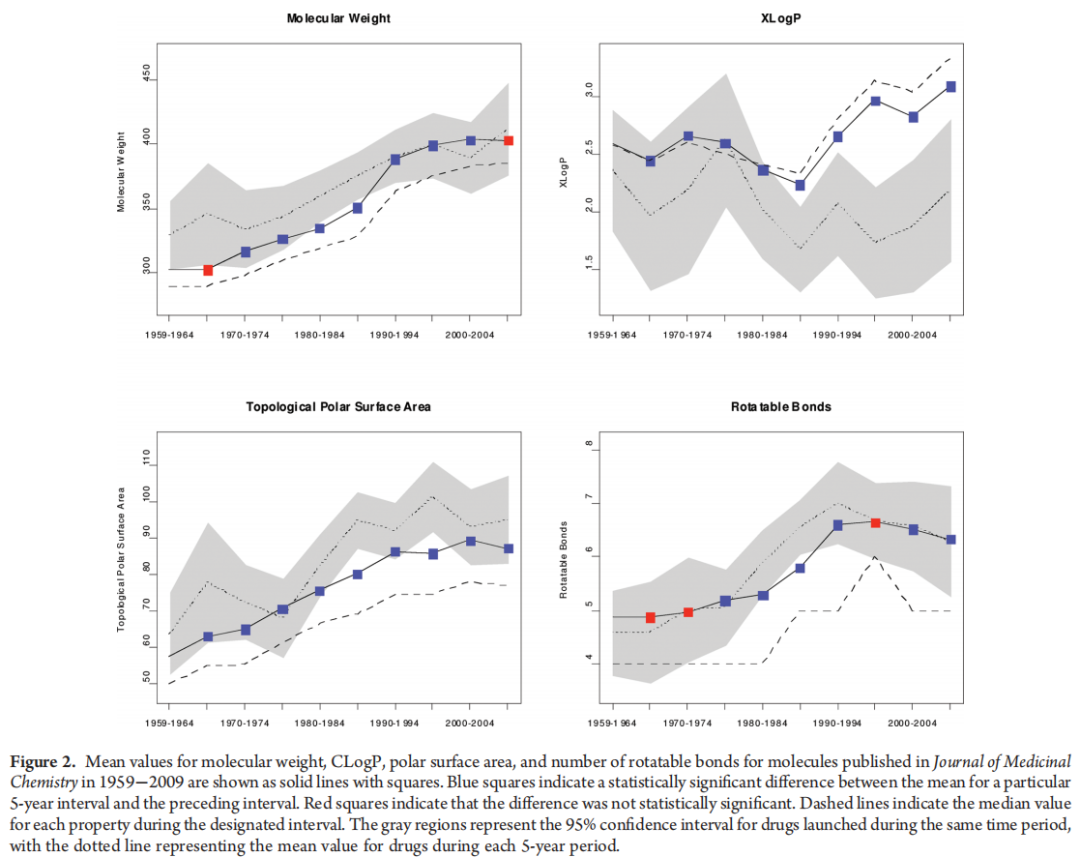
亲脂性是药物设计中的另一个关键因素。亲脂性最常用的替代指标是辛醇-水分配系数或log P。log P的计算预测已研究了40多年,并已成为药物化学实践的常规部分。计算log P(CLogP)值与一系列关键的药物发现参数相关,包括溶解度、渗透性、hERG结合和CNS渗透。研究还指出,亲脂性化合物往往更具混杂性,并且具有更高的非特异性毒性发生率。药物计算log P随时间的变化在近期文献中受到了一些关注。Lipinski报告称,1994年辉瑞药物发现计划中合成的化合物的CLogP比1986年合成的化合物高出14%。
图2显示了与前文分析相同时间区间内药物和JMC发表化合物的CLogP趋势。有趣的是,自1985年以来,JMC发表的化合物的平均CLogP一直高于当前上市药物。1985年之前没有整体趋势,CLogP值在5年区间内上下波动2–4%。从1990年开始,CLogP急剧增加。1990年至1994年间,CLogP中位值增加了17%。随后在1995年至1999年间又增加了10%。2000年至2004年间变化较小(下降了2%),但在本世纪的后五年,CLogP值似乎再次呈上升趋势。然而,由于样本量较小,很难对2005年至2009年这一时期做出明确的评估。
过去十年中,极性表面积(PSA)在药物化学文献以及制药工业中受到越来越多的关注。已经注意到,拥有较大PSA的分子可能在穿越生物膜时遇到困难。这种无法穿越膜的能力可能导致吸收不良或缺乏血脑屏障(BBB)渗透。Palm发表了一些最早将PSA与口服吸收关联起来的研究,他发现高度吸收(>90%)的药物其极性表面积小于60 Ų,而吸收不良(<10%)的药物其极性表面积大于140 Ų。Clark等人通过使用更大的数据集和计算极性表面积的替代方法扩展了这项工作。Egan的后续研究在由log P和PSA定义的性质空间中定义了一个椭圆区域,在该区域内的化合物具有更高的口服吸收概率。这个通过分析《Physician's Desk Reference》中的药物定义的区域或“蛋形”,可以作为化合物设计的目标。PSA与BBB渗透之间的关系最早由van de Waterbeemd于1992年发表,随后Clark等人在后续论文中进一步细化。Hitchcock和Pennington建议将90 Ų作为靶向CNS化合物的截止值,并指出前25名CNS药物的平均PSA为47 Ų。
图2显示,随着时间的推移,药物和JMC发表化合物的平均和中位极性表面积均呈现持续增长。在1959年至1995年间的每个5年区间内,PSA稳步上升,每个区间内中位值增长在2%到10%之间。除少数例外,JMC化合物的PSA值一直处于上市药物观察到的范围之内。
分子柔性是药物发现计划过程中经常优化的另一个性质。药物化学家常常通过刚性化分子来减少构象熵并增加结合亲和力或赋予选择性。可旋转(通常是非环单)键的数量常被用作分子柔性的替代指标。Veber在2002年的一篇论文中将大鼠口服生物利用度与可旋转键数量、极性表面积和氢键基团数量相关联。Lu的后续工作表明,这些关系更为复杂,结果往往因计算方法和所分析分子的治疗类别而异。Hou在2009年的一篇论文通过分析一组706个化合物,发现人类口服生物利用度与可旋转键或任何其他计算参数之间没有关系,对这些结果提出了进一步的质疑。
图2显示了1959年至2009年间JMC发表化合物的平均可旋转键数量有所增加。这些变化与上市药物观察到的趋势平行。平均值从1959年到1990年有所增加,但在过去20年中似乎已趋于稳定。JMC化合物的可旋转键中位数在1965年至1984年间恒定在4。在1985年至1989年期间,数量从4增加到5,在1990年代后期进一步增加到6。
氢键几乎是每个药物发现计划的关键要素。一个恰当放置的氢键可以赋予化合物效力和选择性。然而,像这里讨论的许多参数一样,氢键可能是一把双刃剑。过多的氢键基团会抑制化合物穿越生物膜的能力,并显著降低生物利用度。在1997年的“类药五原则”论文中,Lipinski指出大多数上市药物的氢键供体少于5个,氢键受体少于10个。这些观察结果得到了许多后续研究的支持,并已成为许多药物发现计划的指导原则。
图3显示了1959年至2009年间JMC发表分子中氢键供体和受体的趋势。氢键基团的平均数量在1960年代、1970年代和1980年代有所增加,但在过去20年中似乎已趋于稳定。氢键供体的趋势与上市药物观察到的趋势平行,而受体的平均数量在1980年代和1990年代低于药物观察到的水平。氢键供体的中位数在整个50年期间恒定为1。氢键受体的数量在整个1960年代、1970年代和1980年代早期恒定在3,在1980年代增加到4。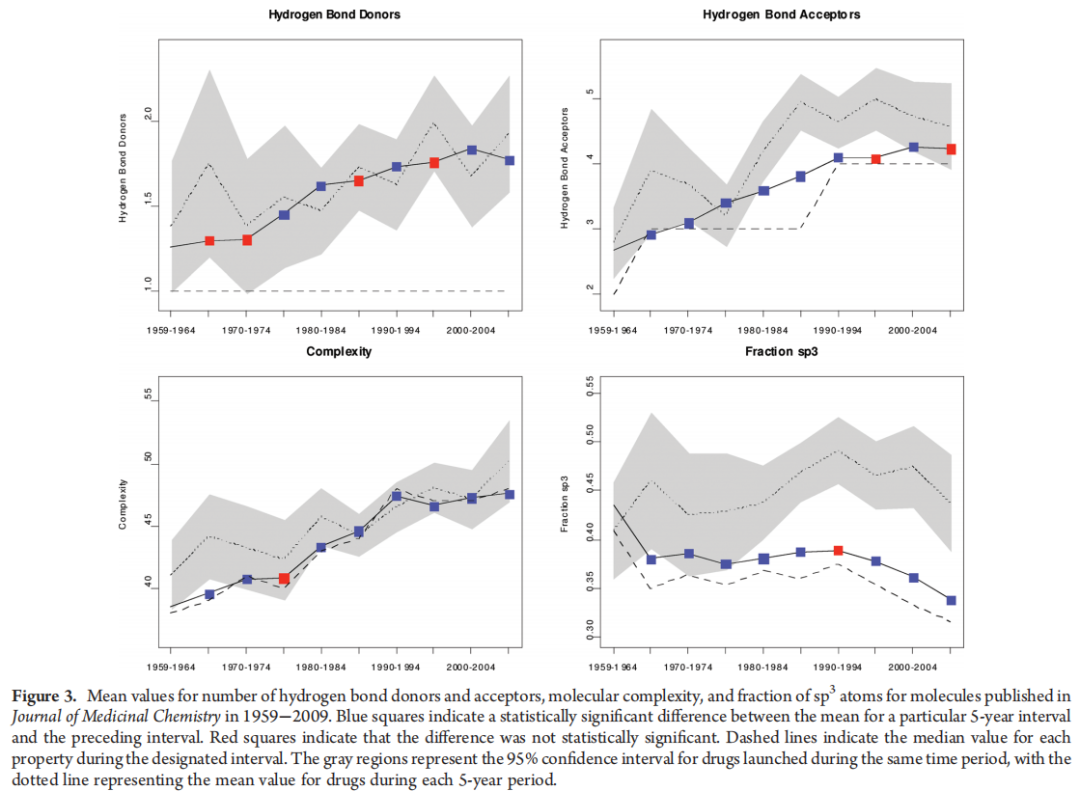
过去50年中,药物化学家们一直在攻克日益具有挑战性的靶点。此外,密切相关的靶点如激酶或离子通道带来了选择性挑战。一种观点认为,更具挑战性的靶点可能需要额外的功能性。在评估药物化学化合物随时间的变化时,我们需要一个指标来比较分子的相对复杂性。我们选择采用了Schuffenhauer在一篇出版物中使用的方法,该方法基于分子指纹中开启的比特数来量化分子复杂性。
图3比较了1959年至2009年间JMC报道分子的复杂性。与此处报告的许多其他参数一样,1959年至1990年间,药物和JMC化合物的复杂性都在稳步增加。JMC化合物的中位复杂性在1980年代有所增加,1980–1985年期间增加了8%,随后的5年期间又增加了5%。JMC复杂性在1990年至1995年间继续增加了10%,但在随后的年份中趋于平缓。然而,或许是由于对选择性和安全性日益增长的需求,2004年至2009年间上市药物的复杂性有所增加。
类药分子的芳香特性近期受到了大量关注。衡量分子芳香特性以及间接衡量其三维形状的一个指标是sp³碳原子比例(Fsp³)。这个最初由Yan和Gasteiger定义的指标,是sp³碳原子数与碳原子总数的比值。Lovering及其合作者在2009年发表的文章表明,Fsp³可以与水溶性和熔点等物理性质相关。作者证明,具有较高Fsp³的分子往往具有较低的熔点和较高的水溶性。这些性质可能会使分子更容易制成制剂。作者还表明,上市药物的Fsp³(0.47)往往高于发现阶段的化合物(0.36)。Ritchie和McDonald最近的研究显示了芳香环数量与溶解度、CYP抑制、血浆蛋白结合和hERG结合等几种性质之间的关系,所有这些都会影响开发化合物固有的挑战。
图3显示了1959年至2009年间JMC发表分子的Fsp³趋势。如图所示,sp³碳原子的中位比例从1960年代中期的0.35增加到1990年代初的0.38。然而,Fsp³在1995年至2009年间稳步下降,在最近测量的时期达到中位值0.32。有趣的是,自1970年代中期以来,JMC发表的分子其sp³特性一直低于上市药物。正如Lovering所述,分子“扁平度”的增加可能是由于促进sp²–sp²偶联的新化学方法所致。
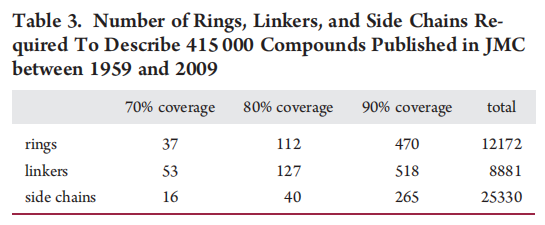
同样有趣的是(表3),JMC中70%的化合物由相对较少的“构建模块”组成:37种环类型、53种连接子和16种侧链。表3中的数据是通过按照Bemis最初描述的方案对分子进行碎片化生成的;环、连接子和侧链按频率排序。然后我们计算了占总数70%、80%和90%所需的环、连接子或侧链的数量。这一发现与早期对已知药物的研究一致。另一方面,有强有力的证据(图4)表明,新分子框架(环+连接子的组合)的引入率近年来实际上有所上升,2007年为1589个,2008年为1935个。换句话说,相同的构建模块正在以新颖的方式组装。有趣的是,新框架的数量似乎与JMC分子中非环-芳香键数量的增加平行。2008年发表的新框架中有25%至少包含一个非环-芳香键。新框架数量的增加可能至少部分是由钯催化偶联反应(如Suzuki、Heck和Negishi反应)的采用所驱动的。
我们对过去50年间《JMC》报道的超过40万个分子的分析清楚地表明,典型分子的性质发生了巨大变化。分子变得更大、更复杂、更具亲脂性,并且根据sp³碳原子比例衡量,它们更扁平、更具芳香性。此外,这些分子更具柔性,它们的极性表面积增加,并且含有更多的氢键供体和受体。毫不奇怪,这些趋势中的许多与上市药物观察到的趋势平行。表4和图6比较了JMC最初5年发表的分子与最近5年期间发表的化合物的计算性质。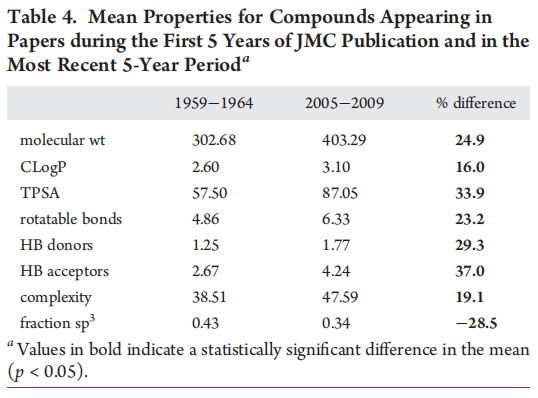
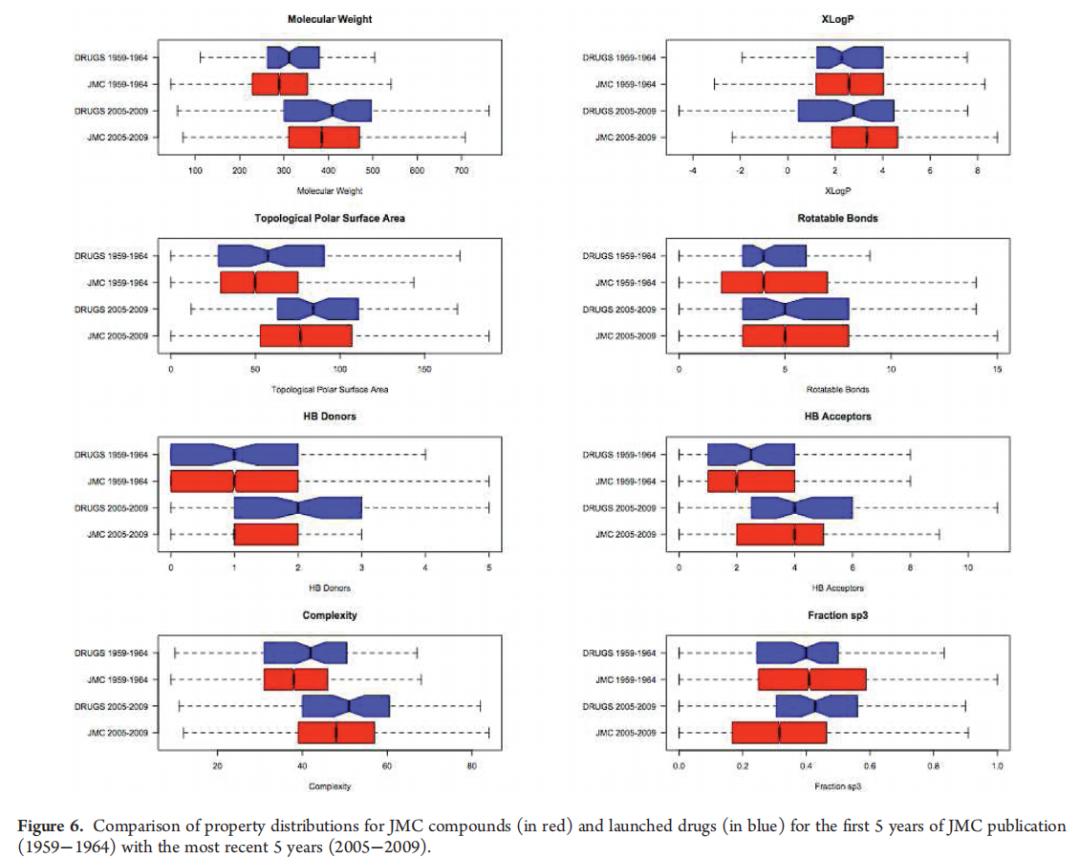
如表4所示,除sp³比例下降了28%外,所有计算性质的平均值增加了16–37%。图6以箱线图形式显示了相同集合的分布。同期上市药物的分布作为参考显示。箱线图中的凹口粗略表示了中位数周围的95%置信区间。如果两个中位数的凹口不重叠,则有强有力的证据表明中位数不重叠。在过去50年中,除sp³比例外,所有性质的中位值均有所增加。同一时期,sp³比例显著下降。进一步检查图6表明,最近5年期间发表的分子的CLogP和sp³比例分布,位于同期上市药物观察到的分布范围之外。
大多数化学家会将这些数据解释为,分子的一些性质正在变得“更差”,或者有时被称为“更不类药”。虽然解释可能有争议,但毫无疑问的是,随着时间的推移,药物和典型JMC分子的CLogP和sp³比例日益分化;也就是说,更高比例的JMC分子的性质落在了成功药物中最常见范围之外。当然,这并不一定意味着这些是“坏”分子;许多成功的药物也落在正常范围之外。
这些趋势几十年来一直很明显,但可能正在减弱。在图2和图3中,大多数曲线在过去15年中似乎已趋于平缓。在表5中,我们比较了1959年至1999年的平均值与2000–2009年的平均值。在除Fsp³之外的每个类别中,近期化合物都具有更高的平均值。与“类药性”背道而驰的其他性质一致,Fsp³下降了。然而,当我们进一步比较最近5年(2005–2009)与前一个5年(2000–2004)时,我们看到许多趋势正朝着“类药”方向回归。但是,必须注意不要过度解读短期趋势。
上市药物性质随时间变化的事实,对我们关于类药性的概念提出了质疑。当Lipinski在1997年发表“类药五原则”论文时,上市药物的平均分子量为369。在最近5年期间,平均分子量为388,增加了6%。我们是应该进行调整以使类药五原则符合当前实践,还是应该采用一个比“类药性”更恰当地描述分子理想性质的术语?
为了更好地理解我们如何重新思考“类药”概念,我们可以思考药物化学家在过去的50年里所合成分子性质变化的九种可能解释。
随着对受体亚型知识的增加,以及对选择性日益增长的关注,基于靶点的反筛选也随之增加。在实践中实现选择性通常需要更大或更复杂的分子。
与1970年代相比,今天制造、纯化和结构表征复杂分子要容易得多。因此,自然地,人们倾向于利用这些改进来制造更复杂的分子。试剂的日益易得也使得合成更大、更复杂的分子变得容易得多。以广泛使用的Suzuki偶联中常见的组分芳基硼酸为例。在过去的15年中,Vertex Pharmaceuticals一直维护着一个庞大的可用试剂数据库。2000年,该数据库包含大约300种市售芳基硼酸。在当前版本的同一数据库中搜索,可得到超过3000种此类试剂。
如上所述,新的sp²–sp²偶联方法的引入,以及这些方法向高通量合成的适应,对药物化学产生了广泛影响。图5显示了1959年至2009年间JMC发表分子中至少含有一个连接两个芳香环的非环单键的分子的比例。过去15年中,带有连接芳香环的非环键的分子比例显著增加,这与“扁平度”的增加(sp³比例下降)是一致的。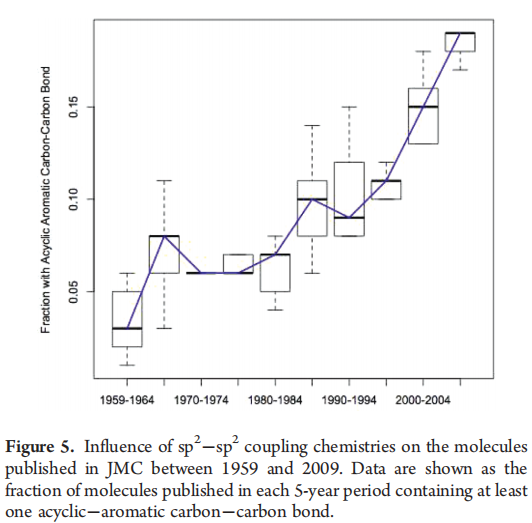
因为制剂科学家经常能够显著改善分子的药代动力学性质,药物化学家可能会倾向于推进次优的化合物,期望物理化学缺陷能在开发过程中被克服。
对疾病分子层面理解的日益应用导致了基于靶点的研究,这促进了性质的顺序优化。例如,发现团队通常先优化受体结合,然后是细胞活性,接着是ADME性质,等等。在这种方法中,很容易陷入局部最优解。在基于靶点的研究兴起之前,更常见的是基于动物功效数据推动项目。这实际上是一种同时进行多因素优化的形式,因为在动物模型中有效的化合物更有可能避免多个问题,例如不溶性、细胞不通透性或代谢不稳定性。
主要来自X射线晶体学的结构信息,常常揭示填充深口袋的明显机会。一种被误导但常见的设计策略只关注效力,会自然而然地增加分子量,更糟糕的是,通常会增加亲脂性,因为这些口袋通常是疏水的。
然而,不应假设基于结构的设计比其他方法更需要对性质膨胀负责。Lipinski在2000年发表的文章显示,无论是“理性”还是高通量筛选(HTS)驱动的药物发现计划所产生的临床候选药物,其包括分子量和亲脂性在内的多项性质都有所增加。更新Lipinski的分析并考察这些趋势在随后的年份是否发生了变化,将是很有启发性的。
在1990年代中期,HTS范式要求构建庞大的化合物库,要么通过从供应商处购买分子,要么通过高通量化学实现。来自高通量筛选的先导分子通常物理性质较差,使优化过程复杂化。
随着时间的推移,对高效力、选择性等参数的要求不断提高,这推动了分子复杂性的增加,并常常降低了“类药性”。
为了完整起见,我们也考虑了这些变化也可能源于JMC编辑政策变化的可能性。JMC发表分子数量的增长显著超过新药数量的事实可能表明,近年来,早期阶段的项目以及优化程度较低的化合物更有可能在JMC上报道。
如果《JMC》是“制药化学家制造了什么”的合理替代指标,那么我们已经确认了以下事实:在过去50年中,分子广泛物理性质的平均值和中位数一直在稳步或周期性跃升。图2和图3总结了1959年至2009年间JMC发表分子的性质。亲脂性(以XLogP衡量)和三维性(以Fsp³衡量)都正在偏离成功药物中通常发现的范围。该期刊报道的几种性质(特别是分子量和log P)的上行趋势似乎在1985年左右有所加速。这提供了一个有价值的线索:显然,性质变化并非仅由高通量化学和筛选导致,因为这些技术直到1990年代中期才变得普遍。同样,分子复杂性的增加也不是最近的现象,因此不能完全归因于组合化学和高通量筛选等因素。
值得注意的是,这些趋势似乎在1980年代中期加速,表明在1980年代早期发生了某种变化。对1980年代早期(在组合化学或高通量筛选时代之前)性质上升趋势最可能的解释似乎是分子生物学的进步,即对受体亚型的理解导致了对特异性的关注;基于靶点的药物设计及其相应的逐项性质优化范式(可能因结构生物学而加剧);以及使得合成和表征更复杂分子成为可能的技术改进。
近年来,人们对天然产物和类似、更复杂的分子作为药物发现计划的起点重新产生了兴趣。正如Keller所指出的,许多天然产物违反类药五原则但仍然具有生物利用度。这些例外的原因尚不清楚,但进一步的研究应能让我们定义有价值的新规则。
尽管人们感知到对“类药性”的强调,但制药产出持续下降。截至2009年10月,药物批准数量比上一年下降了29%,比10年平均水平低了13%。如果JMC报道的化合物,正如我们所主张的,是对药物发现计划中正在生产的分子类型的合理反映,那么看来我们关于“类药性”的许多认识并未被始终如一地付诸实践。更准确地说,尽管有许多出版物概述了开发性质超出明确范围化合物的挑战,但我们发现,药物化学家实际制造的分子正越来越多地成为这样的异常值,原因如上所述的许多合理且可理解的理由。
展望未来,我们认为有必要为化合物设计选择一套不同的统一原则。我们将越来越多地被迫研究前所未有的复杂靶点,同时面临日益增加的安全性和有效性障碍。简单的规则,即使被广泛采用,也将不够。回归1950年代和1960年代普遍存在的、仅依赖动物药理学作为主要读数的做法似乎不太可能。相反,我们提出五个相互依存的原则来改进设计过程:
(1)更深入地理解真正的类药性。简单的数值经验法则仅在一定程度上有所帮助。相反,我们需要更深入地理解,尽管某些分子的物理性质超出通常范围,但什么使它们具有类药性。这些规则可以是全局的(适用于所有分子)或局部的(针对每个化学类别或靶组织进行微调),对于攻克“不可成药”靶点尤其有价值。
(2)更多地强调配体效率,特别是在苗头化合物到先导化合物的过程中。这将反过来支持对筛选库的重新评估,或许包括基于片段方法的使用。更高的配体效率也往往会具有更低的log P,这已被明确证明可以减少毒理学风险。
(3)更具信息量和更高通量的体外ADME/毒性试验。试验技术的进步使得药物发现团队越来越多地采用额外的体外试验。药物发现计划中的化合物现在常规进行溶解度、CYP和hERG试验测试。Caco-2和平行人工膜渗透等体外系统常被用作渗透性的替代指标。已经开发了许多细胞系统来指示潜在的不良结果。虽然许多体外试验有益处,但与体内数据的相关性可能不一致。体外试验质量和适用性的提高将使药物化学家能够更早地获得洞察并避免潜在风险。
(4)回归同步多变量优化的思维方式。药物发现计划的最终目标是产生一种对人类安全有效的化合物。为了开发这样的化合物,团队必须优化许多标准,包括亲和力、选择性、活性、性质和药代动力学。这些标准常常以串行方式优化。团队会优化单个标准(如酶效力),然后牺牲这些成果去优化第二个性质(如溶解度)。用一个性质换取另一个性质的过程可能重复数十个周期。可以采用多种方法来支持多目标优化。一种是使用可视化软件,使团队能够理解数据的全貌。一些研究组最近开发了面向药物发现计划的可视化工具。另一种方法是应用多目标优化算法,尝试同时优化多个标准。虽然多目标优化已成功应用于许多其他领域,但在药物发现中的主要限制是计算模型的准确性有限。
(5)减少对“简单”化学的依赖。虽然钯介导的sp²–sp²偶联和酰胺键形成反应有其地位,但我们相信,在药物化学中更强调合成艺术将极大地改善我们分子的物理性质。
采纳这些原则将具有挑战性,但我们相信,如果药物化学要维持其活力和生产力,方法上的重大变革十分重要。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢