
JAK2是多种炎症性疾病、癌症以及类风湿性关节炎的重要治疗靶点。抑制JAK2已成为治疗上述疾病一种极具前景的策略。2026年1月27日,全北国立大学的研究人员在《Acta Pharmacologica Sinica》上发表文章,题为“AI and experimental convergence: a synergistic pathway to JAK2 inhibitor discovery”。

该研究计算了多种分子描述符,并基于这些特征构建了机器学习模型。在所有模型中,CatBoost结合Morgan指纹的表现最佳,在测试数据集上取得了0.94的准确率。随后,利用该CatBoost模型对韩国化学数据库(KCB)进行了虚拟筛选,以识别最具潜力的JAK2抑制剂。为评估排名靠前分子的性能,开展了多种计算分析。最终筛选出四种化合物进行实验验证,结果表明其IC₅₀值均小于10 μM。将AI驱动的建模与实验验证相结合,为个性化医疗提供了一种极具前景的策略,有助于开发更加精准和高效的激酶靶向治疗药物,同时显著降低新药进入临床试验所需的时间和成本。
代码仓库:
https://github.com/MaryamRasoolSatti/jak2
背景
JAK蛋白是一类参与细胞信号转导的胞内酪氨酸激酶。JAK家族包含四种同工型:JAK1、JAK2、JAK3和TYK2,它们约在30年前由Wilks等人首次发现。四种JAK亚型均可级联激活七类STAT蛋白,并触发转录因子的激活,从而调控约40种不同细胞因子和化学信号分子的基因表达,以调节细胞功能(图1)。因此,JAK蛋白作为免疫相关疾病的潜在治疗靶点,受到了广泛关注。然而,JAK家族成员之间的ATP结合位点在结构上高度相似,使得通过传统方法为特定JAK亚型筛选具有高选择性的配体面临较大挑战。
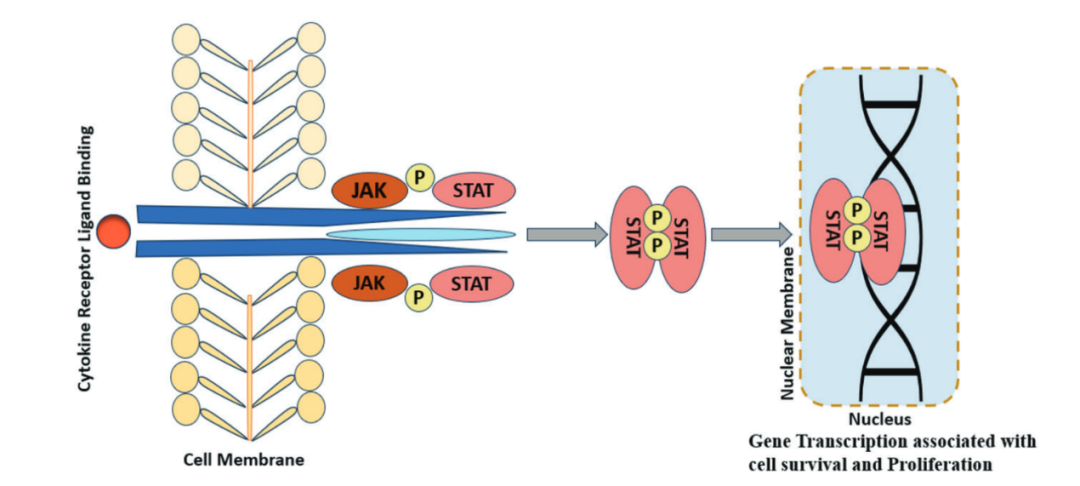
图1 JAK2在细胞因子信号传导中的作用机制:激活、磷酸化及信号转导
JAK2被认为是炎症性疾病、癌症及类风湿性关节炎的潜在诊断标志物和治疗靶点。在过去几十年中,多种靶向JAK2的药物已进入临床或临床前研究阶段,用于治疗骨髓增殖性疾病,其中部分药物已获得美国食品药品监督管理局(FDA)及其他监管机构的批准。开发具有更优药物相容性和成药性质的新型JAK2抑制剂,仍具有重要研究价值和应用前景。
方法
首先进行数据集构建,从ChEMBL数据库中整理生物活性数据,并依据IC₅₀阈值进行标注(图2a)。然后计算分子指纹(Morgan、PaDEL、MACCS),并与生物活性标签配对,作为机器学习模型输入(图2b)。对于集成学习模型训练阶段,采用Butina聚类方法对数据进行聚类,并通过五折交叉验证进行超参数优化和模型验证(图2c)。其中,训练集以及测试集如表1所示。随后,对化合物进行虚拟筛选、分子对接和聚类分析;排名靠前的分子进一步接受分子动力学模拟、ADME性能评估及抑制活性实验,最终获得具有预测JAK2抑制活性的优选候选物(图2d)。
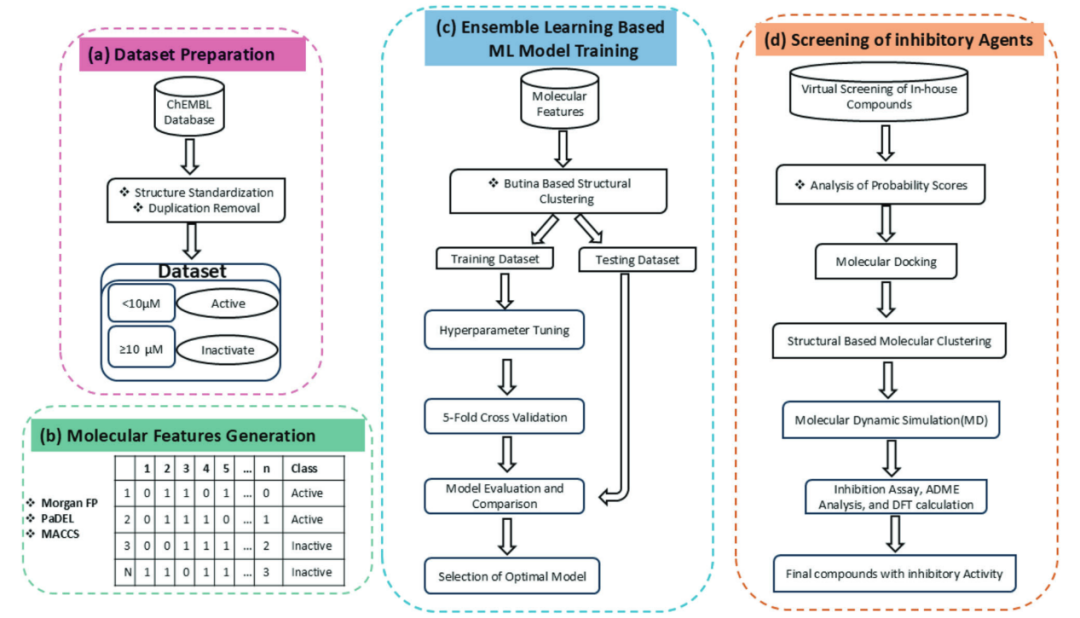
图2 JAK2抑制剂预测流程
表1 用于构建JAK2机器学习模型的数据集概况

结果
数据集解析
为构建机器学习模型,从ChEMBL数据库收集了与JAK2蛋白相关的抑制剂与非抑制剂数据集。采用非线性降维方法t-SNE探索分子集合所覆盖的化学空间。在此之前,基于分子的SMILES表达式计算了长度为1048位的Morgan指纹。随后,使用主成分分析(PCA)对特征空间进行降维,以降低维度并减少噪声干扰。结果显示,抑制剂在化学空间中的分布趋于集中,而非抑制剂则分布在不同区域(图3a)。抑制剂与非抑制剂在空间分布上的明显差异表明,其分子结构特征存在显著不同,这种结构差异可能是导致其生物活性差异的重要原因。
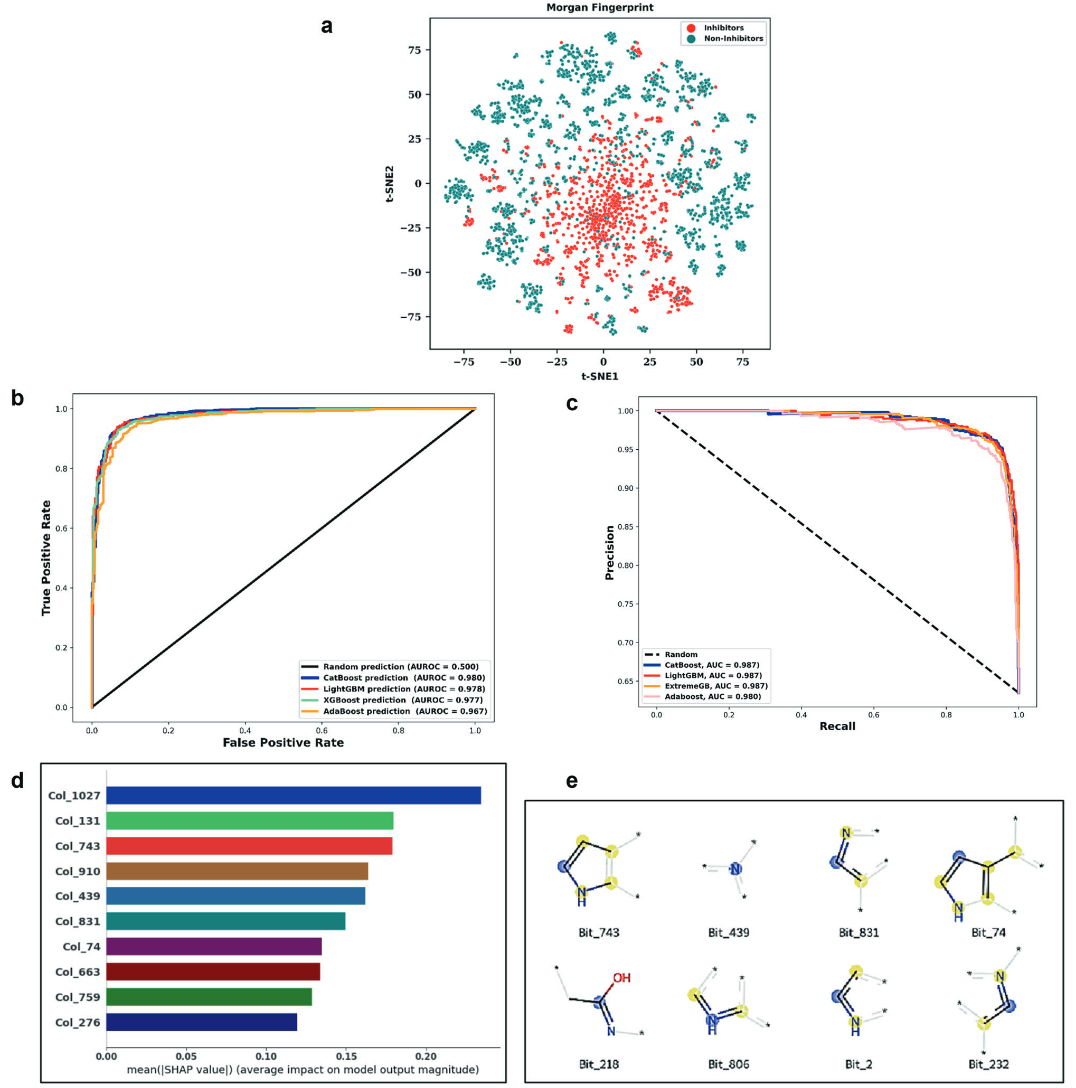
图3 数据集抑制剂与非抑制剂分布以及机器学习模型性能
机器学习模型性能评估
本研究评估了多种机器学习模型的预测性能。五折交叉验证结果表明,在所有分类器中,CatBoost表现出最为稳健的预测能力(表2)。
表2 基于Morgan指纹的五折交叉验证数据集性能
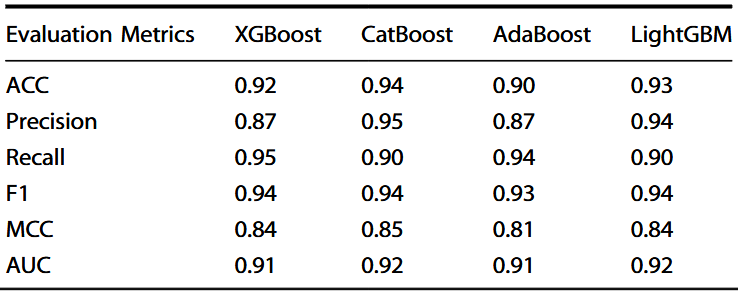
在其他评价指标方面,如Matthews相关系数(MCC),CatBoost取得了0.86的最高得分,明显高于其他模型,表明其在区分JAK2抑制剂与非抑制剂方面具有更强的判别能力。在测试数据集上的评估进一步验证了CatBoost的优越性,其准确率为0.94,MCC为0.86(表3)。
表3 基于Morgan指纹的测试数据集性能
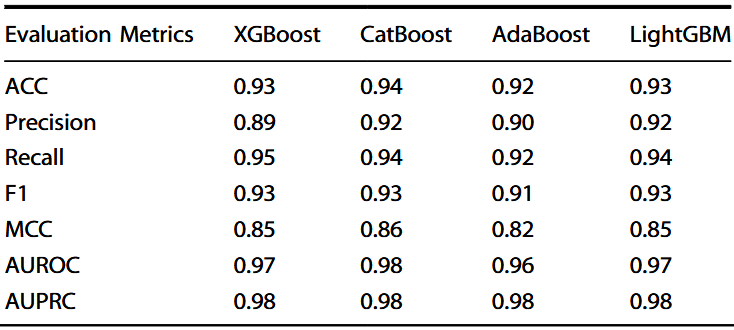
此外,测试集的ROC曲线显示CatBoost的AUROC达到0.980,高于XGBoost、LightGBM和AdaBoost,表明其具有极强的区分能力(图3b)。PR曲线分析结果显示,CatBoost、LightGBM和XGBoost的AUPRC均为0.987,而AdaBoost为0.980(图3c)。综合多项评价指标可以看出,尽管多种模型均表现良好,但CatBoost在各项性能指标上始终优于其他模型,是预测JAK2抑制剂最为可靠的分类器。
分子描述符对模型性能的贡献
采用基于SHAP的特征重要性分析方法,识别对测试集预测最具影响力的分子特征。影响力由高到低的前十个特征如图3d所示。其中,Morgan指纹位点[1027, 131, 743, 910, 439, 831, 74, 663, 759, 276]被认为是最关键的结构片段位,表明这些结构在分子中的存在对其抑制活性和结合强度具有显著贡献。
进一步分析了最重要特征及其对应的分子子结构片段,以探究其与JAK2结合活性的关系。结果显示,这些子结构位点是影响JAK2蛋白结合位点抑制剂生物活性的关键因素(图3e)。其中,位点743对应小分子中的吡啶环,该结构有助于与结合位点残基形成氢键相互作用;位点439属于氨基官能团,强调了在药物–靶标相互作用过程中维持分子酸碱性质的重要性;位点74表示甲基吡咯烷结构,对维持靶蛋白结合所需的几何构型和构象取向至关重要;位点218对应分子中的乙酰胺基团,该基团在药物化学中应用广泛,尤其常见于抗癌和抗炎药物中。上述结果表明,所构建的机器学习模型能够有效识别与JAK2抑制活性密切相关的关键分子子结构特征,从而实现对高效JAK2抑制剂的精准预测。
基于机器学习的虚拟筛选
KCD包含约73万多个小分子化合物,被用于高通量虚拟筛选。在虚拟筛选之前,对数据库进行了预处理,包括筛选具有类药性的小分子、去除盐形式分子,以及利用RDKit库生成互变异构体和构象。经预处理后,共获得53万多个小分子,并按照与机器学习模型训练阶段相同的方法,计算其1048位Morgan指纹。随后,将处理后的小分子特征作为输入,导入由已知抑制剂数据训练得到的CatBoost分类模型进行预测,并根据预测概率对分子进行打分。最终,从模型预测结果中筛选出预测概率大于0.5的3474个化合物,作为潜在的JAK2抑制剂候选分子。
分子对接分析
通过虚拟筛选获得的候选分子被用于分子对接分析,以研究其在JAK2结合位点的结合强度及构象取向。所有筛选得到的分子均被对接至JAK2蛋白的活性口袋中,其最高Glide评分达到−8.010 kcal/mol。为进一步严格筛选KCB数据库中的候选分子,选取了结构多样化分子的最优构象,并使用Canvas工具中的k-means方法,基于结构相似性进行聚类分析。最终形成14个聚类,并从每个聚类中依据较高的Glide评分选取一个代表性小分子。随后,对这14个分子进行诱导契合对接,以更精确地分析受体蛋白和配体的柔性变化。其中,JAK2-C3的Glide评分最高,为−11.471 kcal/mol(图4)。
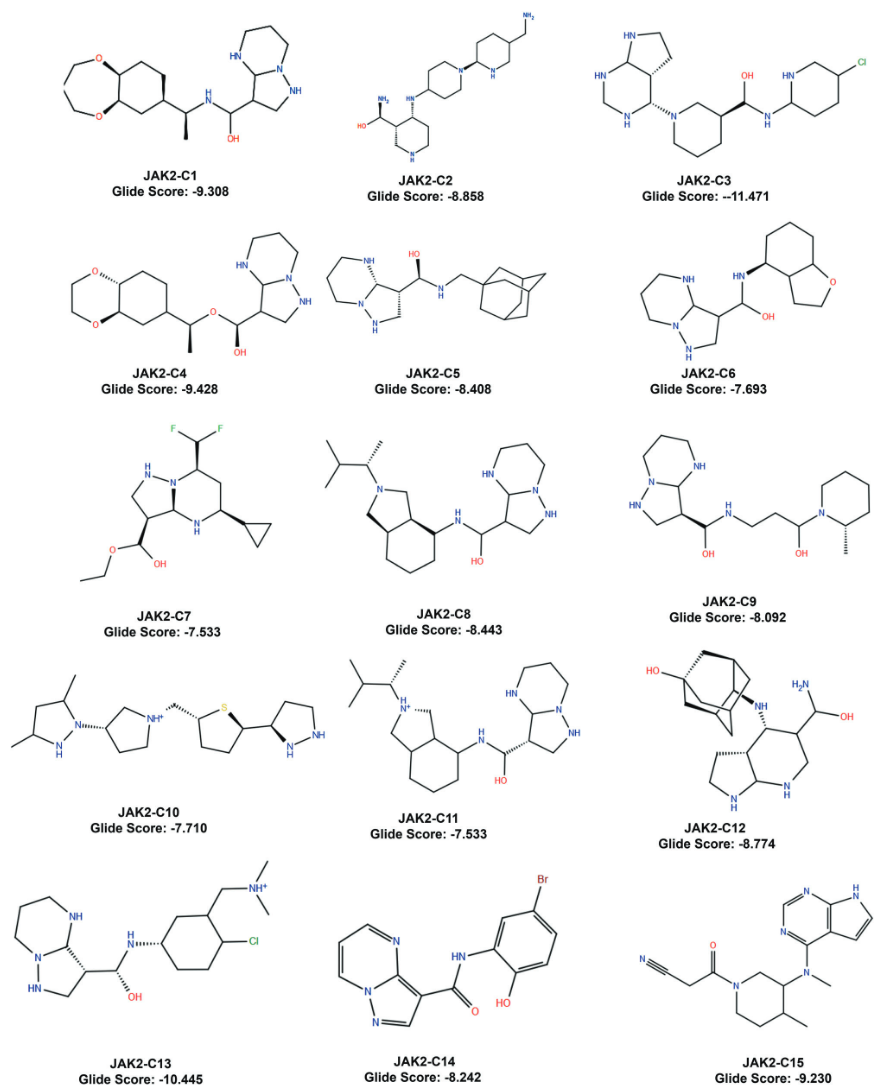
图4 虚拟筛选与分子对接后筛选出的候选化合物的化学结构及其Glide评分
分子动力学模拟与结合自由能分析
为评估候选分子的结构稳定性,进行了MD模拟分析。以蛋白主链C-α原子为参考绘制RMSD曲线(图5),结果显示,在初始平衡阶段(约20 ns)后,JAK2-C1、JAK2-C2、JAK2-C4和JAK2-C13复合物的RMSD波动较小,表现出较为稳定的构象。这些复合物的稳定RMSD值维持在1.5–2.5 Å之间,表明其具有典型的蛋白–配体动力学稳定性。作为对照,tofacitinib在整个模拟过程中RMSD始终低于2Å,显示出更高的稳定性,为比较候选分子的结合强度提供了参考基准。
回转半径Rog分析进一步揭示了JAK2蛋白–配体复合物的整体紧凑性和结构稳定性。如图5a所示,所提出分子的Rog值在约19.0–21.5 Å的狭窄范围内波动,表明在不同配体结合过程中,蛋白的三级结构整体保持稳定。其中,JAK2-C1、JAK2-C2、JAK2-C4和JAK2-C13复合物的Rog值变化最小,显示其在模拟过程中维持了良好的结构紧凑性和稳定性。
此外,RMSF分析用于评估JAK2蛋白–配体复合物中各残基的柔性变化。结果显示,在蛋白核心区域,尤其是活性位点附近,RMSF值普遍低于2 Å,表明该区域在模拟过程中保持较高的刚性和稳定性;而在N端和C端区域,RMSF值则升高至3–5 Å,反映出末端区域具有较大的构象柔性(图5a)。JAK2-C1、JAK2-C2、JAK2-C4和JAK2-C13复合物的RMSF峰值分布较为均匀,说明这些分子未引起明显的局部扰动,能够维持蛋白活性位点及其周围残基的整体稳定性,进一步支持其作为潜在先导化合物的可行性。
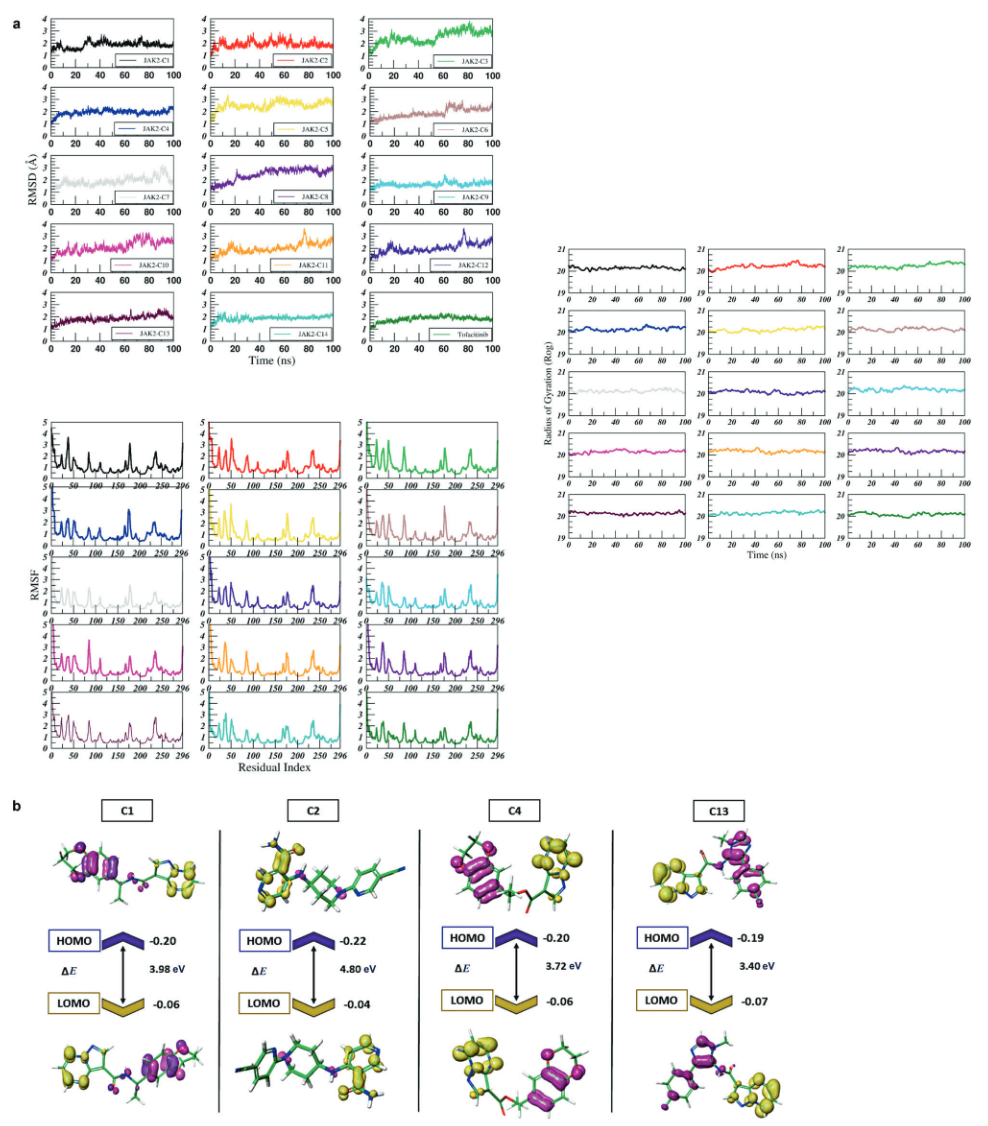
图5 MD模拟分析以及JAK2顶级抑制剂的HOMO与LUMO分析结果
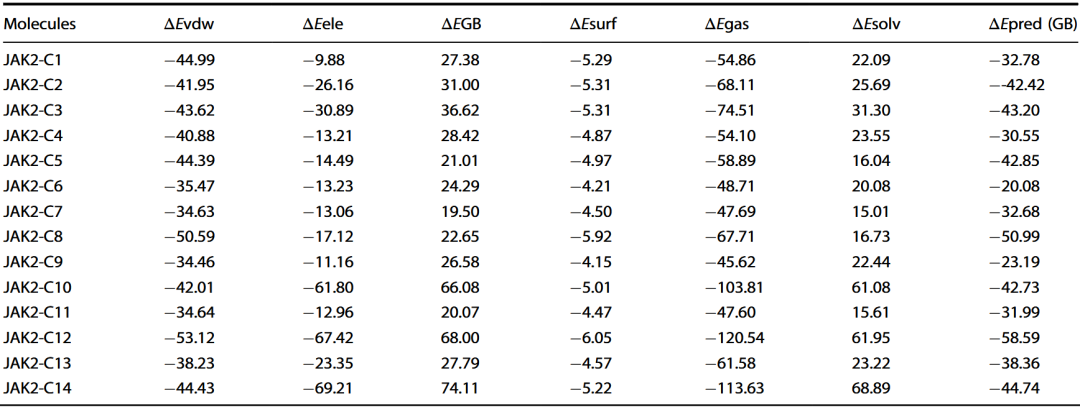
采用MMPBSA方法对筛选出的JAK2候选分子进行了结合自由能计算。结果表明,JAK2-C12 的结合自由能最为有利;其次为JAK2-C1、JAK2-C2、JAK2-C4和JAK2-C13(表4)。这些能量结果表明,上述分子在JAK2结合口袋中具有稳定的相互作用模式及良好的极性接触,有利于形成稳定的蛋白–配体复合物。
表4 JAK2苗头化合物的结合自由能计算结果
密度泛函理论计算
对分子对接筛选出的高排名候选分子进行了DFT计算,以分析其轨道能级、化学反应性及动力学稳定性。DFT结果显示,JAK2-C1具有最低的能隙,表明其具有较高的化学反应活性;而JAK2-C2表现出最高的电负性,说明其具有更强的吸电子能力(表5)。
表5 DFT方法计算的JAK2抑制剂的HOMO能级、LUMO能级、能隙及电负性

先导分子的HOMO–LUMO分布如图5b所示,其中蓝色区域代表分子中电势较正的区域,红色区域则表示电势较负的区域。JAK2-C13具有最小的能隙(3.40 eV),显示其在JAK2结合位点可能具有较高的反应活性;而JAK2-C2的能隙最大(4.80 eV),且其HOMO能级最低,表明该分子在电子结构上更为稳定,但反应活性相对较低。
ADME性质分析
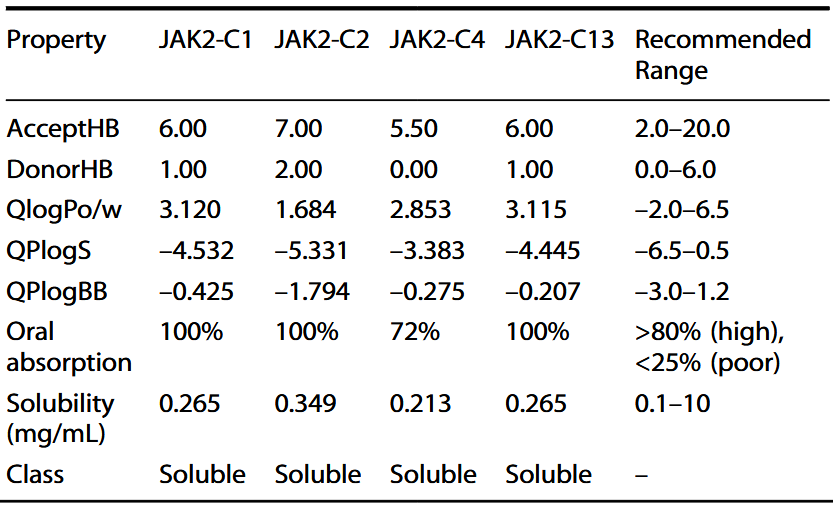
对经过MD模拟筛选出的四个候选分子进行了ADME性质评估。结果表明,所有候选分子均符合常规类药性规则,即氢键供体数不超过5,氢键受体数不超过10,且QlogPo/w值小于5。预测的水溶性(QPlogS)均处于安全范围内。血脑屏障参数显示合理的数值,表明这些分子不太可能对中枢神经系统产生不良影响。此外,预测结果显示所有候选分子均具有良好的口服吸收能力和适宜的溶解度与渗透性。因此,这些候选分子的药代动力学性质均处于理想范围内,使其成为具有潜力的选择性JAK2抑制剂(表6)。
表6 所筛选JAK2抑制剂的计算机模拟ADME性能分析结果
JAK2抑制实验评估
采用剂量–反应分析方法,对筛选得到的四个最优化合物及阳性对照药物tofacitinib的JAK2激酶抑制活性进行了评估。实验结果表明,所有候选化合物均表现出显著的JAK2抑制活性。其中,JAK2-C13显示出最强的抑制效果,其次分别为JAK2-C4、JAK2-C1和JAK2-C2(图6)。
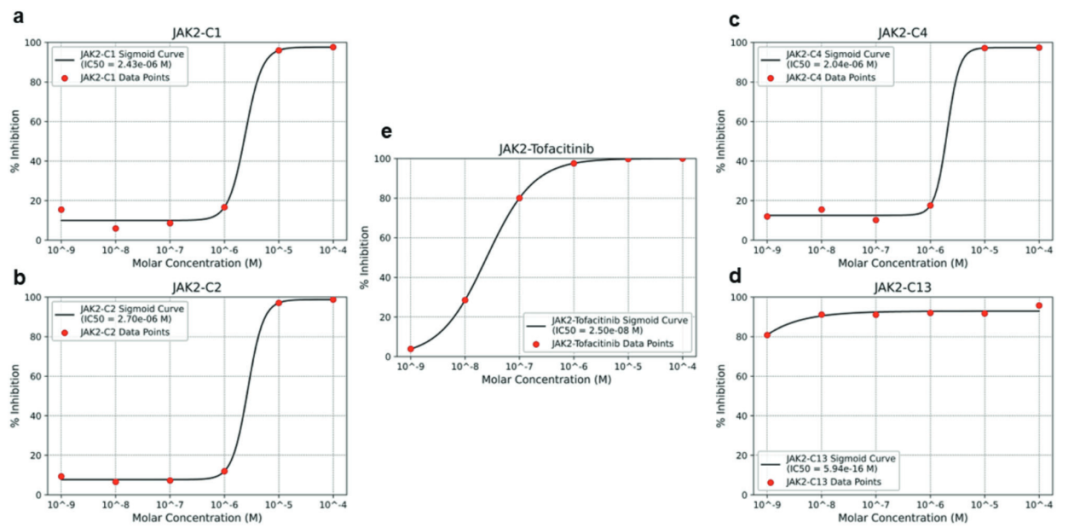
图6 JAK2激酶抑制活性的剂量–反应曲线
未来方向
未来的研究可着重于提升AI模型的可解释性,并进一步探索其在预测脱靶效应和毒性特征方面的应用,以推动该策略向更高水平发展。此外,将AI驱动模型与实验验证相结合,为个性化医疗开辟了新的研究方向,有助于开发更加精准、高效的激酶靶向治疗方案,同时显著降低新型治疗药物进入临床试验所需的时间和经济成本。
参考链接:
Maryam, Cho, H., Pokhrel, A. et al. AI and experimental convergence: a synergistic pathway to JAK2 inhibitor discovery. Acta Pharmacol Sin (2026).
https://doi.org/10.1038/s41401-025-01701-9
--------- End ---------
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢