
DRUGONE
研究人员提出了一种基于几何向量感知器图神经网络的模型,用于直接从原子坐标预测承诺函数,从而绕过传统需要人工构造集体变量的步骤。该方法具有原子级可解释性,能够自动识别在复杂分子转变中起关键作用的原子,而无需预设反应坐标。该模型在多种分子体系上测试后均能准确推断承诺函数,并量化每个重原子在转变机制中的重要性,同时还能精确估计相关过程的速率常数。整体而言,该方法通过实现无需集体变量的学习以及自动识别物理上合理的反应坐标,为理解复杂分子动力学过程提供了新的计算工具。

在复杂分子体系中,稀有跃迁过程通常通过转变路径理论进行分析,其中承诺函数描述了从某一构型出发先到达产物态而非反应态的概率,因此被视为理想的一维反应坐标。传统计算承诺函数的方法包括路径采样和变分策略,而近年来机器学习逐渐用于从集体变量构造复杂反应坐标。然而这些方法仍依赖预先选择的集体变量,这些变量通常由物理直觉给出,例如原子距离或二面角,但这种选择缺乏定量标准,且低维变量往往难以充分描述复杂转变。尽管已有研究尝试直接从笛卡尔坐标学习集体变量,但计算成本较高。因此研究人员提出直接从完整原子坐标学习承诺函数,从根本上避免显式集体变量的依赖。
方法
研究人员构建了一个基于GVP-GNN的承诺函数网络。首先从分子动力学轨迹中提取构型,并将其表示为分子图,其中原子作为节点、相互作用作为边,同时包含标量与向量特征。随后通过几何向量感知层对节点和边进行等变变换,并通过消息传递机制传播结构信息。模型输出为每个构型对应的承诺值,并通过基于时间相关函数的变分原理构建损失函数进行训练。在最终阶段,通过对节点嵌入进行置换不变的聚合操作得到图级预测,从而学习承诺函数映射。研究人员还通过敏感性分析计算每个原子对预测结果的重要性,实现原子级可解释性。
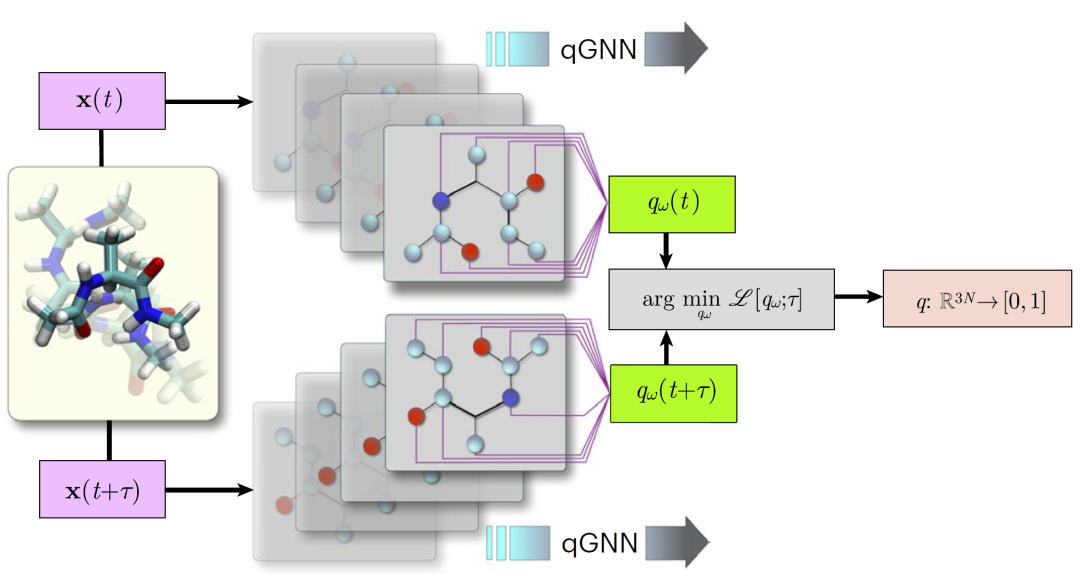
图1:qGNN学习流程。
结果
NANMA构象异构化
研究人员首先在经典测试体系NANMA上验证方法。模型从原子坐标学习得到的承诺函数在投影到二面角空间后与已有数值方法结果一致,并清晰给出分界面位置。节点敏感性分析显示,决定主二面角的关键原子在预测中具有最高重要性,这表明模型能够自动识别主导反应坐标的结构自由度,而无需显式提供这些变量。
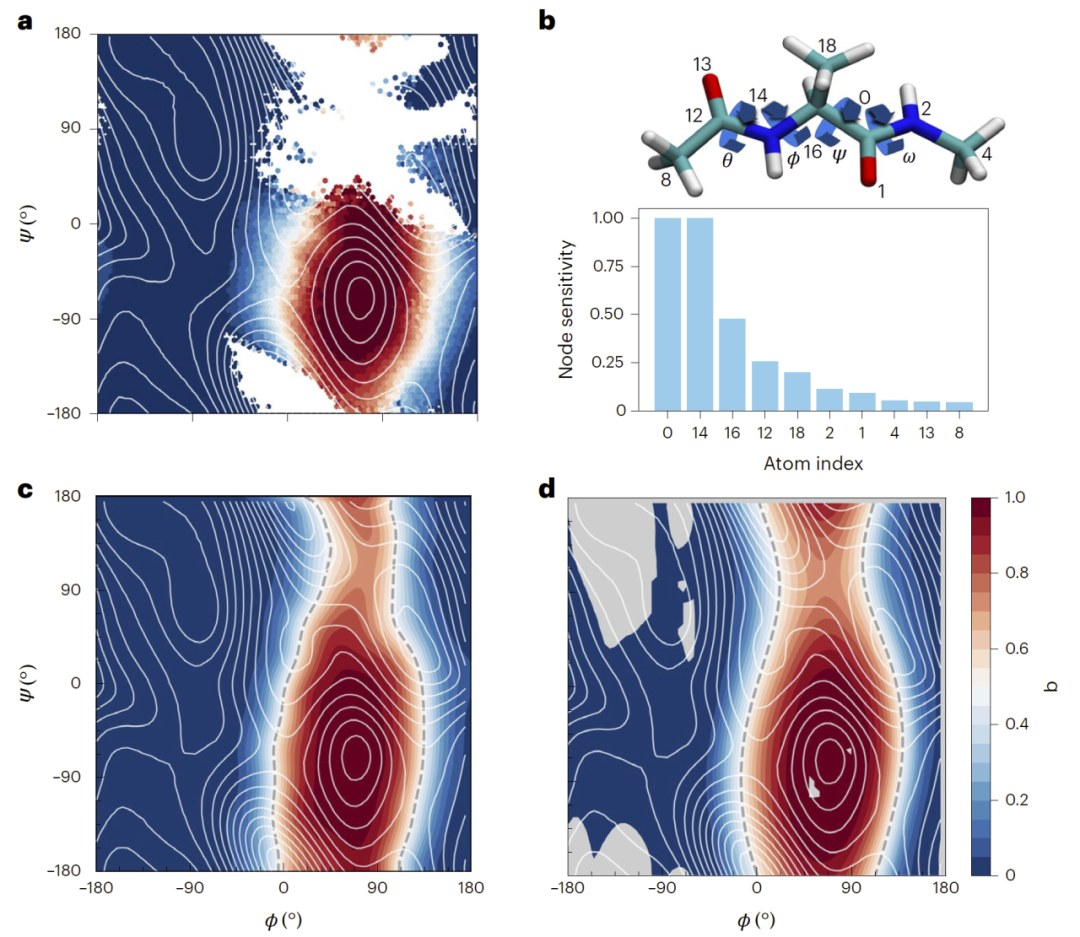
图2:NANMA分子异构化过程。
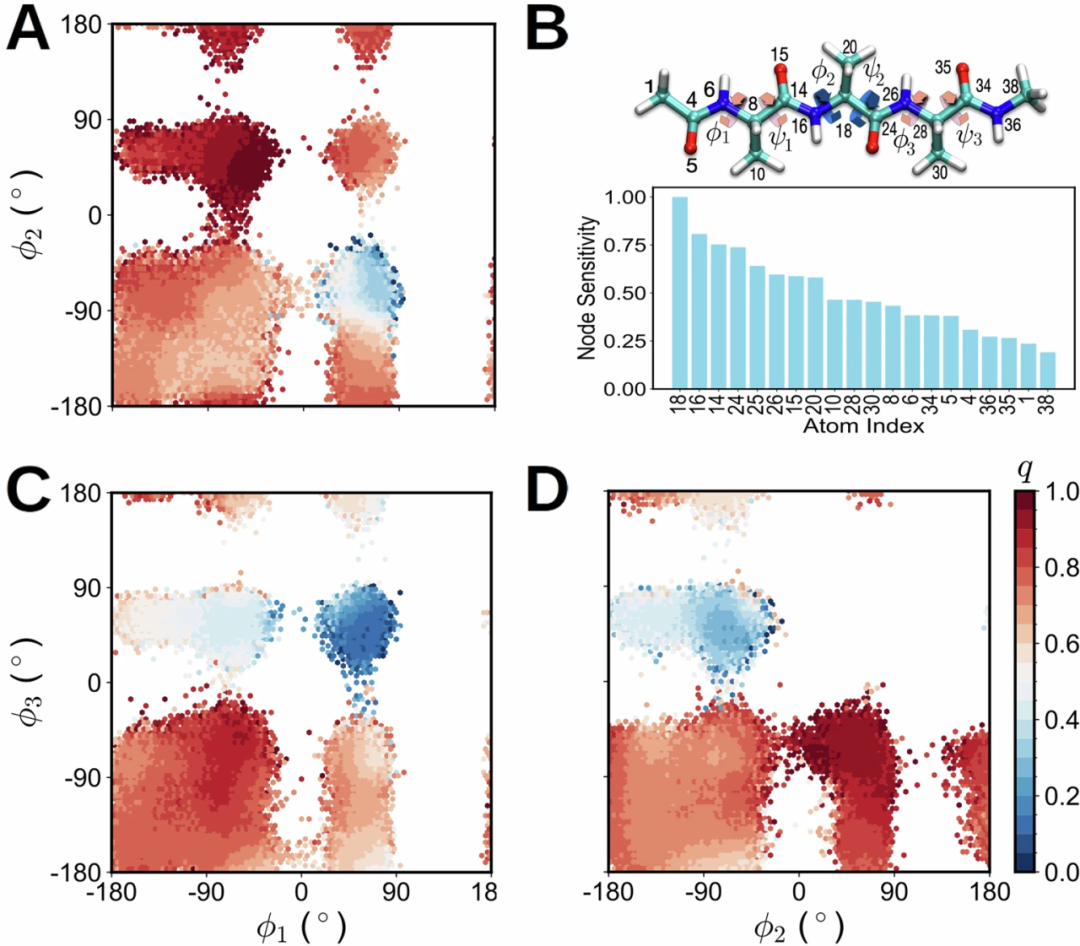
三丙氨酸构象平衡
在更复杂的三丙氨酸体系中,模型同样成功恢复承诺函数分布,并识别三个主链二面角对应的关键原子组。敏感性分析进一步证实这些扭转角是驱动构象转变的主要自由度,说明该方法能够在更高维体系中稳定识别动力学主变量。
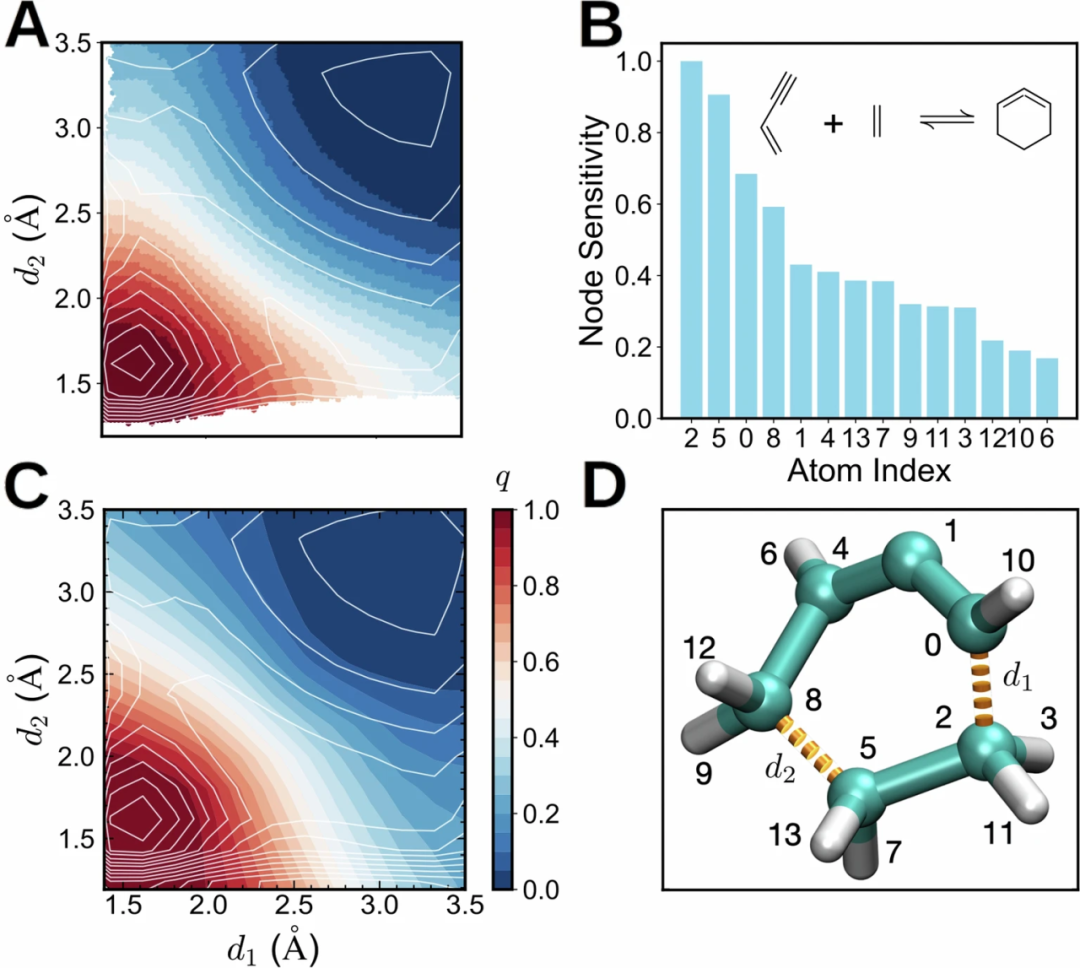
Diels–Alder反应
研究人员随后将模型应用于有机化学中的Diels–Alder环加成反应。结果显示模型预测的承诺函数分界面与理论研究一致,并成功识别参与新键形成的碳原子为最关键节点,同时还发现其他相关原子在结构重排中具有重要作用。这说明该方法不仅适用于构象变化,也适用于真实化学反应过程。
Trp-cage蛋白折叠
在更接近真实生物体系的Trp-cage小蛋白折叠任务中,研究人员利用长时间分子动力学轨迹训练模型。尽管缺乏参考承诺函数,模型在RMSD和端到端距离空间中仍呈现清晰的折叠与展开分界。敏感性分析识别出与蛋白折叠关键距离相关的重要残基位置,并通过聚类分析发现过渡态结构呈现二级结构部分丢失而两端保持稳定的特征。此外在更大的villin蛋白体系中,模型同样能够直接从原子坐标学习承诺函数并准确估计转变速率,证明其具有良好可扩展性。
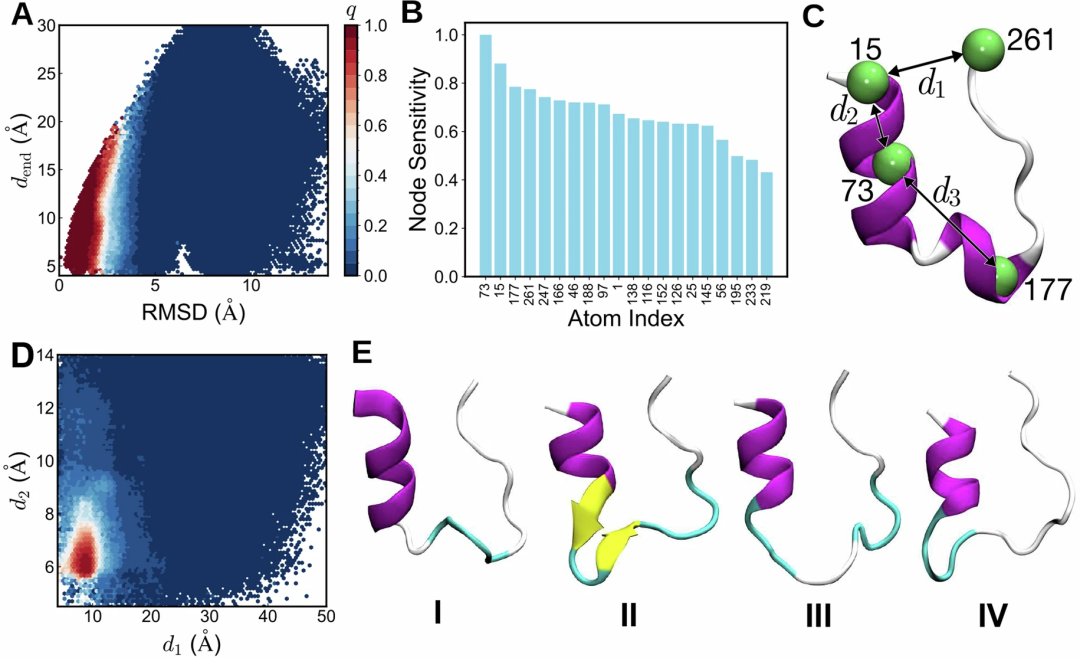
讨论
研究人员指出,该GVP-GNN模型能够在无需人工设计集体变量的情况下直接从原子坐标学习承诺函数,同时捕捉空间几何与相互作用关系。与传统基于低维变量的模型相比,这种方法避免了降维带来的信息损失,并允许直接在完整坐标空间分析过渡态结构。节点敏感性分析还能自动识别最关键原子,为构建可解释反应坐标提供依据。尽管该方法仍依赖能够覆盖稀有转变的充分采样轨迹,但它提供了一种灵活的坐标空间建模框架,可用于推断物理可解释变量,甚至直接作为增强采样的集体变量。未来该框架还可扩展到多稳态体系,并与马尔可夫状态模型或自适应采样策略结合,从而更系统地研究复杂分子动力学过程。
整理 | DrugOne团队
参考资料
Contreras Arredondo, S., Tang, C., Talmazan, R.A. et al. Learning the committor without collective variables. Nat Comput Sci (2026).
https://doi.org/10.1038/s43588-026-00958-2

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢