
DRUGONE
在真实条件下预测大规模原子体系的光电性质对于材料设计至关重要,但基于第一性原理的量子力学计算在大体系中计算成本极高,难以应用于实际材料开发。近年来,神经网络方法在材料建模中展现出潜力,但通常需要大量训练数据,并且缺乏物理可解释性。相反,基于物理模型的近似方法虽然更高效,但往往精度不足且难以推广。
研究人员提出一种物理约束的哈密顿学习框架 HAMSTER,通过在近似物理模型基础上引入机器学习,对复杂体系的量子力学哈密顿量进行预测。该方法只需少量第一性原理计算即可捕捉环境变化对电子结构的影响,并能够在不同温度、不同组成以及大尺度结构中准确预测光电性质。研究结果表明,该框架可以处理包含数万原子的体系,为大规模材料模拟提供了新的可扩展解决方案。

量子力学计算在材料科学、量子生物学和药物设计等领域具有重要作用,尤其是在光电材料研究中,需要考虑缺陷、热扰动和结构无序等真实条件,这些因素会显著影响电荷输运和复合过程。理想情况下,应直接在有限温度下对大规模体系进行电子结构计算,但传统第一性原理方法通常假设理想晶格和零温条件,难以描述真实材料中的复杂行为。
机器学习力场的发展使得分子动力学模拟能够以较低成本达到第一性原理精度,但这些方法通常只能预测原子运动轨迹,而无法直接提供电子结构信息。在许多情况下,仍需要密度泛函理论计算电子结构,而该方法随体系尺寸增大计算成本急剧增加,限制了其在大规模体系中的应用。
近年来,研究人员开始探索通过机器学习直接预测哈密顿量的方法,从而避免重复进行第一性原理计算。哈密顿学习的核心思想是训练模型从原子结构预测哈密顿矩阵,再由该矩阵计算能带、带隙和其他电子性质。然而,这一任务具有挑战性,因为哈密顿量不仅取决于原子结构,还依赖于所选基组和表示方式。为提高泛化能力,研究人员引入满足对称性的等变神经网络,使模型在旋转和平移变换下保持一致性,从而提高数据效率。
尽管如此,现有方法仍需要大量训练数据,并且难以描述复杂材料在有限温度下的动态行为。因此,需要一种结合物理模型和机器学习的新方法,以在保证精度的同时提高可扩展性。
方法
研究人员提出的HAMSTER框架基于物理启发的近似模型,并结合机器学习对其进行修正。首先使用简化的物理模型描述体系的基本电子结构,该模型能够捕捉主要物理效应,但精度有限。随后,机器学习模型学习第一性原理计算与近似模型之间的差异,从而得到更准确的哈密顿量。
模型输入为原子结构及其局部环境,通过等变神经网络预测哈密顿矩阵的修正项,使结果满足对称性要求。由于模型建立在物理近似之上,因此只需要少量第一性原理数据即可训练完成。
训练完成后,模型可以用于分子动力学轨迹中的每一个结构,从而在有限温度下计算电子结构和光电性质。该方法不仅降低计算成本,还能够描述温度、缺陷和化学组成变化对材料性能的影响。
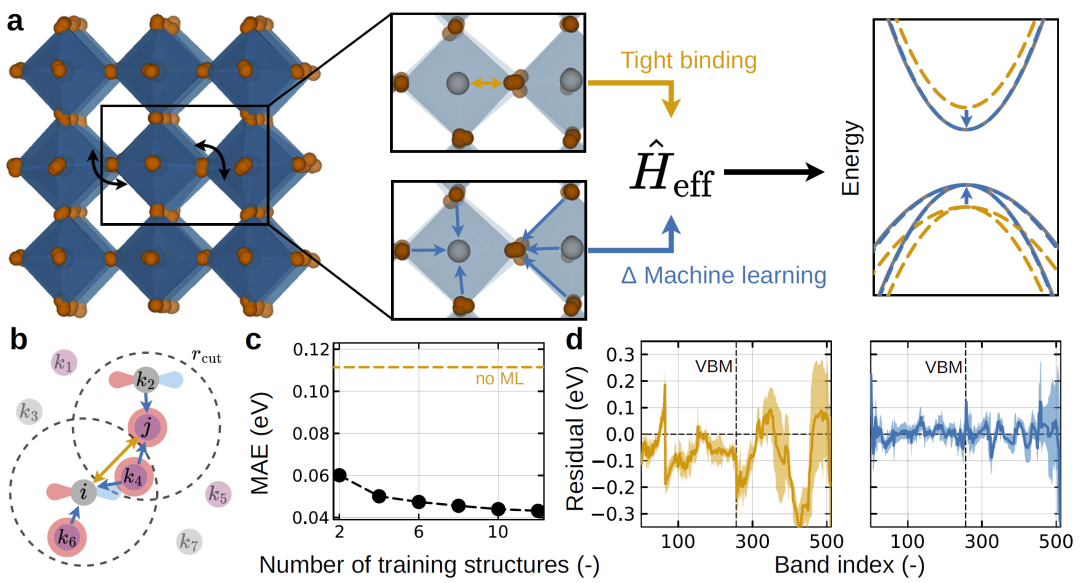
图1:物理约束哈密顿学习模型与整体流程。
结果
大规模体系中的哈密顿预测
研究人员首先验证模型在卤化物钙钛矿体系中的表现。这类材料具有强烈的热扰动和结构无序,对电子性质影响显著。结果表明,HAMSTER能够在不同结构尺寸下准确预测哈密顿量,并保持与第一性原理结果一致。
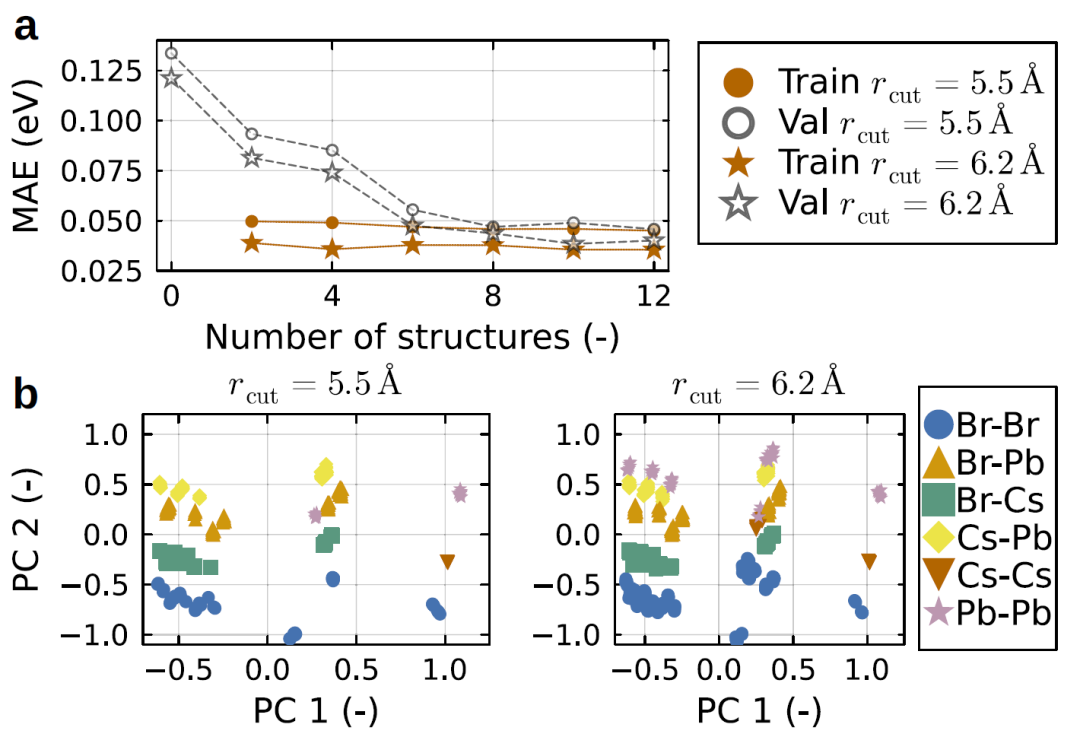
图2:CsPbBr₃ 体系的模型训练过程与描述符分析。
温度依赖光电性质预测
在分子动力学轨迹上计算电子结构时,模型能够捕捉温度变化对带隙的影响,并与实验结果一致。相比传统方法,该框架可以在更大的体系中进行计算,并保持较高精度。
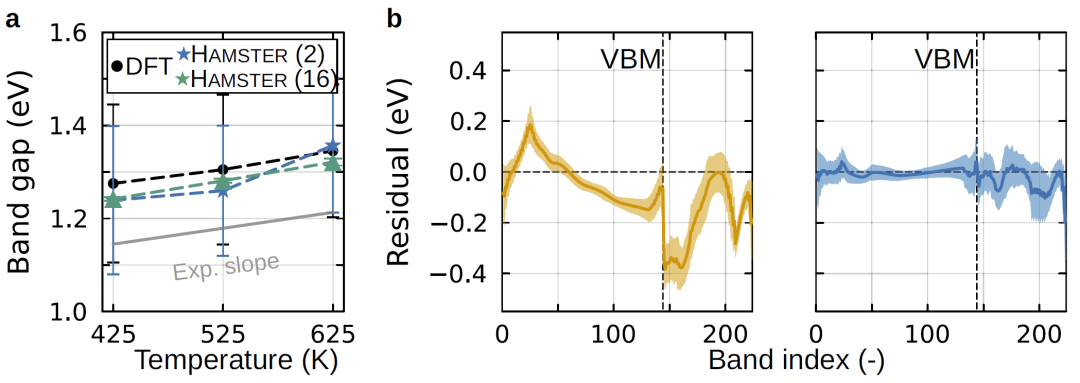
图3:模型在不同温度条件及大规模体系计算中的可迁移性。
计算效率与可扩展性
研究人员进一步分析模型的计算复杂度,发现该方法的计算成本随体系尺寸近似线性增长,而传统第一性原理计算呈立方增长。因此,HAMSTER可以处理包含数万原子的体系,而传统方法难以实现。
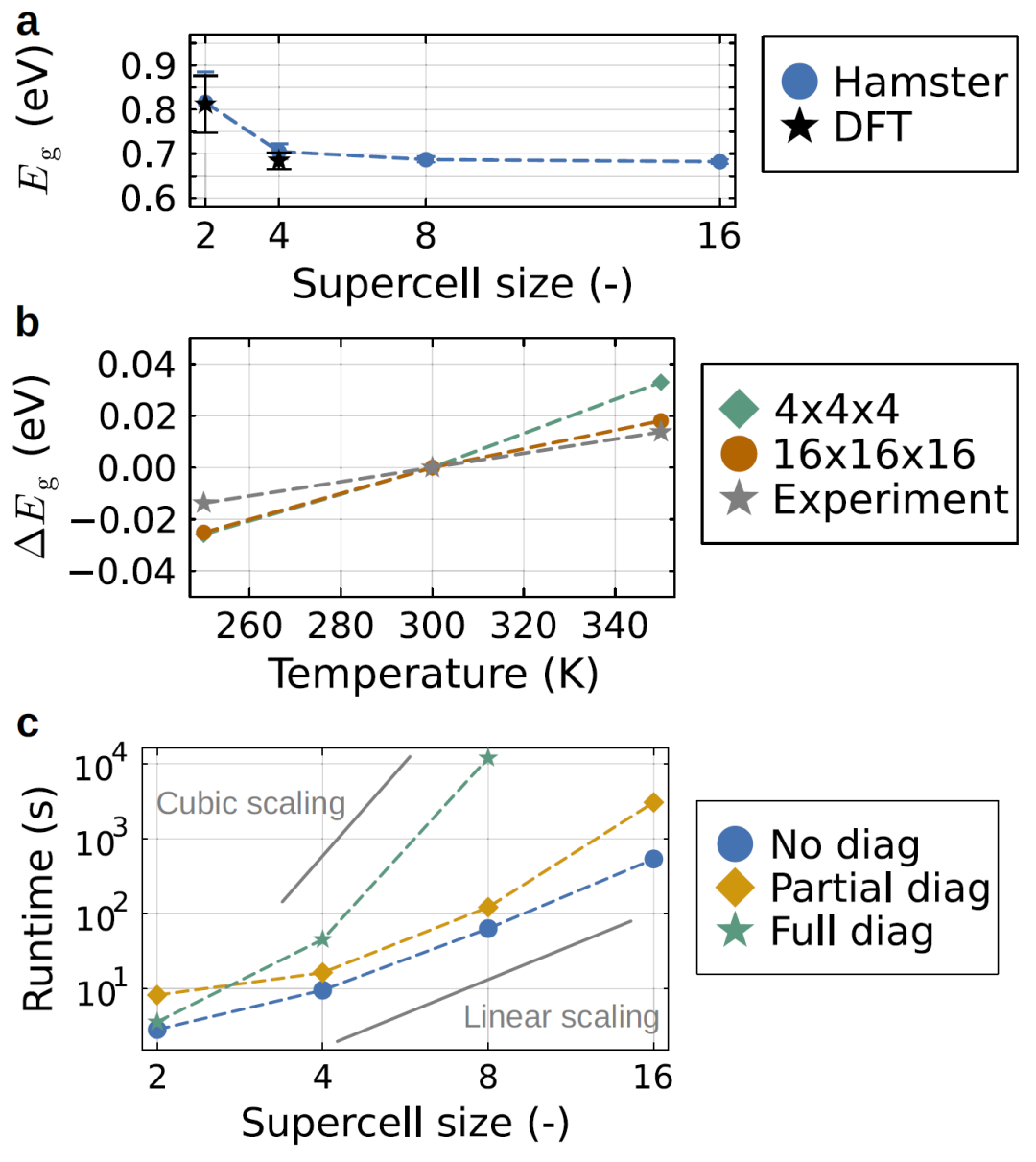
图4:MAPbBr₃ 在不同超胞尺寸和温度下的带隙变化及计算规模随体系大小的扩展关系。
复杂环境下的电子结构预测
在含缺陷和不同组成的材料中,模型仍能保持较高精度,说明物理约束的哈密顿学习具有良好的迁移能力。这种能力对于真实材料模拟尤为重要,因为实际材料通常包含复杂的局部环境。
讨论
本研究提出的物理约束哈密顿学习框架实现了在大规模体系中预测电子结构和光电性质的可行方案。通过结合近似物理模型与机器学习,研究人员在保证精度的同时显著降低计算成本,并提高了模型的可解释性。
该方法能够在有限温度和复杂环境下进行量子力学建模,突破了传统第一性原理方法在体系规模上的限制。研究人员认为,这种物理驱动的机器学习策略是未来材料建模的重要方向,特别适用于需要同时考虑结构无序、热效应和化学变化的光电材料。
未来工作将进一步扩展该框架,使其能够处理更多材料类型,并与更大规模的分子动力学模拟结合,从而推动高性能光电材料和功能材料的设计。
整理 | DrugOne团队
参考资料
Schwade, M., Zhang, S., Vonhoff, F. et al. Physics-informed Hamiltonian learning for large-scale optoelectronic property prediction. Nat Commun 17, 2652 (2026).
https://doi.org/10.1038/s41467-026-70865-7

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢