

前瞻性发现高效选择性 BTK 共价抑制剂
原文信息
Shamir Y, Gabizon R, Rogel A, Lin DY, Andreotti AH, London N.
Discovery of Covalent Ligands with AlphaFold3.
Journal of the American Chemical Society, 2026.
DOI: 10.1021/jacs.5c22222
通讯作者:Nir London,魏茨曼科学研究所化学与结构生物学系
摘要速读
本文报道了一项里程碑式工作:将 Google DeepMind 的全原子结构预测模型 AlphaFold3(AF3) 首次系统性地应用于共价配体的虚拟筛选(covalent virtual screening),并在前瞻性筛选实验中成功发现了结构全新的 BTK 共价抑制剂。
核心贡献如下:
1. COValid:首个专门用于共价虚拟筛选富集分析的基准数据集(874 个活性分子 + 37,919 个性质匹配诱饵分子) 2. 方法论突破:AF3 的 mPAE 指标在全部 10 个靶点位点均大幅优于传统共价对接工具(71.8% vs ~10%,Adjusted LogAUC) 3. 实验验证:前瞻性筛选发现 YS1,IC₅₀ = 30 nM(体外),107 nM(细胞),晶体结构验证预测精度达 0.50 Å RMSD 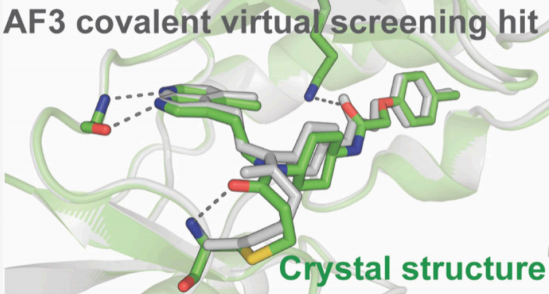
一、研究背景与科学问题
1.1 共价抑制剂的重要性
共价抑制剂(covalent inhibitors)通过与靶蛋白上的亲核氨基酸残基形成不可逆(或可逆)共价键来发挥作用。相较于非共价抑制剂,其优势包括:
• 更高效力:持续占据靶点,即使药物浓度降低后仍保持活性 • 更长靶点占用时间(target engagement):不受药动学波动影响 • 潜在的选择性优势:可利用特定位置的半胱氨酸等氨基酸进行精准定向
截至目前,FDA 已批准超过 50 种共价药物,近年代表性品种包括:
1.2 共价虚拟筛选面临的挑战
共价结合是一个两步动力学过程:
E + I ⇌ E·I → E-I(共价加成物)
KI kinact其中:
• 第一步:可逆非共价识别(KI,由分子识别决定) • 第二步:共价键形成(kinact,由反应活性与空间朝向决定)
传统共价对接软件(DOCKovalent、DOCK6、AutoDock 等)存在以下根本性缺陷:
1. 只对已成键的加成物状态(adduct state)打分,完全忽略共价反应前的过渡态空间朝向 2. 无法量化亲电试剂的固有反应活性,导致多弹头共价库(multi-warhead library)的排序不可行 3. 使用固定蛋白骨架,难以处理配体诱导的构象变化(induced-fit effects) 4. 缺乏标准化富集评估基准:非共价筛选领域有 DUD-E、DUDE-Z 等成熟基准,共价领域从未有过
正是这一空白,催生了本文的核心工作。
二、COValid——首个共价虚拟筛选富集基准数据集
2.1 数据集设计原则
COValid 的构建借鉴了非共价领域 DUD-E 的两大核心原则:
原则 1:物化性质匹配(property matching)
诱饵分子与活性分子在以下 6 个性质上保持高度匹配,避免因性质差异导致的人为富集:
• 分子量(MW) • 氢键供体数(HBD) • 氢键受体数(HBA) • 可旋转键数(rotatable bonds) • 净电荷(net charge) • 脂水分配系数(cLogP)
原则 2:拓扑不相似性(topological dissimilarity)
通过打乱分子骨架连接方式生成诱饵,确保其与活性分子组成相似但拓扑不同,从而排除因化学相似性导致的假阳性富集。诱饵骨架均选自 ZINC20(商业可及化合物数据库),保证化学合理性。
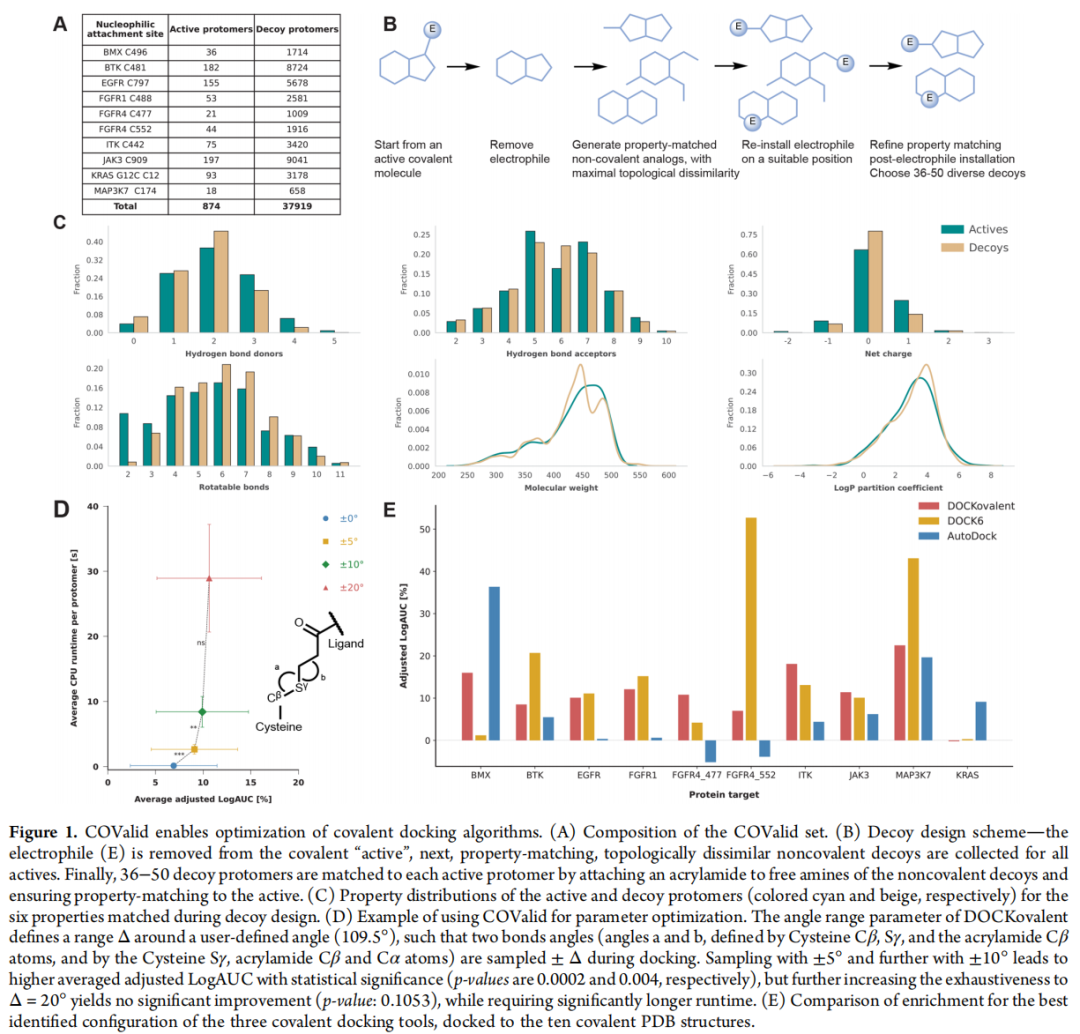
2.2 数据集组成
| 合计 | 10 个位点 | 874 | 37,919 |
注:本研究聚焦于丙烯酰胺(acrylamide)弹头——最常见的靶向半胱氨酸的共价弹头,兼顾实用性与可比较性。
三、AlphaFold3 共价共折叠(Covalent Cofolding)方法
3.1 AF3 的输入与输出
AF3(2024 年 11 月发布)首次实现了全原子生物分子结构预测,支持共价蛋白-配体复合物的预测。
输入:
• 蛋白质氨基酸序列 • 配体的几何优化 3D 构象(SMILES → 3D) • 共价键指定:配体原子编号 + 蛋白侧链原子
输出:
• 预测的共价复合物三维坐标 • 多种置信度指标矩阵(PAE、pTM、ipTM 等)
相比传统对接的关键优势:
• 无需晶体结构,仅需蛋白序列 • 允许蛋白骨架柔性(backbone flexibility) • 可建模配体诱导的构象变化(如 BTK P-loop 重排) • 可暴露隐蔽口袋(cryptic pockets)
3.2 关键指标:mPAE(最小预测对齐误差)
PAE(Predicted Aligned Error)矩阵是 AF3 输出的结构置信度矩阵,其中元素 (i, j) 表示:以 token i 为参考对齐时,token j 位置的预测误差(单位:Å)。
研究者定义:
即:以所有配体原子为参考对齐时,所有蛋白残基位置预测误差的最小值。
mPAE 相较其他置信度指标的优势:
| mPAE | 极高 | 最优 |
mPAE 的物理意义:反映配体与蛋白之间最紧密接触区域的结构置信度,本质上是对结合界面几何互补性的度量,而非简单的全局置信度。
3.3 mPAE 的绝对值与结合概率
在 COValid 适用范围内,mPAE 的绝对值具有前瞻性筛选意义:
| 96.6% | |
这一单调递减关系为设定筛选截断值(cutoff)提供了理论依据。
四、基准测试结果
4.1 COValid 上的富集性能对比
富集度量指标:Adjusted LogAUC
• 0%:随机水平 • 85.5%:理论最优(所有活性分子均排在所有诱饵之前) • 强调早期富集(early enrichment),更贴近实际筛选需求
| AF3 + mPAE | 71.8 ± 5.9% | ~266 s |
值得注意的细节:
• AF3 + Rosetta 重打分(纯物理打分,不依赖 AI 置信度指标)已显著优于传统对接,说明 AF3 生成的结构模型物理合理性足够好 • mPAE 在此基础上进一步大幅提升(28.5% → 71.8%),说明 AF3 内置的置信度信息具有独立的预测价值 • 各位点 AUC(ROC 曲线下面积)范围:0.9082 ~ 0.9996,几乎接近完美分类
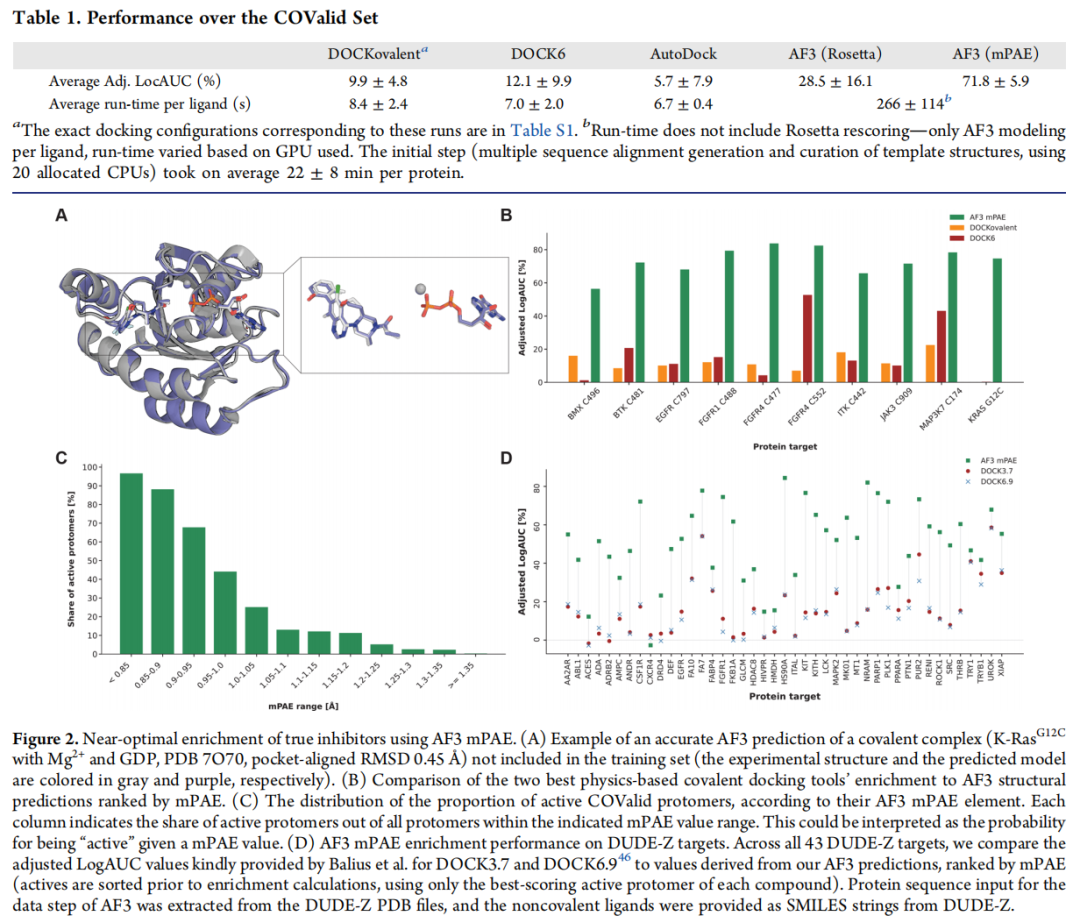
4.2 消融实验:排除训练数据泄露的可能
潜在偏差:AF3 的训练集包含大量 PDB 结构,若测试化合物与训练集重叠,则富集性能可能被高估。
研究者的消融策略:按 Tanimoto 相似性(Morgan 指纹,radius=2,2048 bits)过滤与 PDB 中任何分子相似的活性化合物(及其对应诱饵),在 Tc = 0.4 严格截断下(过滤掉大多数活性分子后),富集性能几乎不受影响。
此外,64 个"实验诱饵"(ChEMBL 中被测试过但活性 > 10 μM 的丙烯酰胺化合物)的 mPAE 均值为 1.5 ± 0.5 Å,与活性分子形成显著区分(86% 的实验诱饵 mPAE > 0.95 Å)。
4.3 非共价虚拟筛选:在 DUDE-Z 上的表现
将 AF3 + mPAE 推广到非共价领域(DUDE-Z 基准,43 个靶点):
• 在 42/43 个靶点中,AF3 显著优于 DOCK3.7 和 DOCK6.9 • 激酶靶点平均 Adjusted LogAUC:60.7 ± 11.1%(优于非激酶靶点的 47.5 ± 21.3%) • 与 Boltz-2(另一 AI 共折叠模型)性能相近
五、前瞻性筛选:发现 BTK 全新共价抑制剂
5.1 筛选流程
靶点:BTK(布鲁顿酪氨酸激酶)Cys481,B 细胞恶性肿瘤的重要治疗靶点,已有 Ibrutinib、Zanubrutinib、Acalabrutinib、Rilzabrutinib 等多个 FDA 批准的共价抑制剂。
筛选流程:
构建虚拟库(~906K 含丙烯酰胺化合物)
↓
AF3 预测各化合物与 BTK Cys481 的共价复合物结构
↓
按 mPAE 排序,保留 mPAE < 0.9 Å 的化合物(440 个,~0.05%)
↓
过滤与已知 BTK 抑制剂相似的化合物(Tc > 0.35)
↓
聚类分析 + 人工审查(多样结合模式)→ 390 个候选
↓
合成 13 个代表性化合物
↓
实验评价(LC/MS、激酶抑制、细胞实验、晶体学)5.2 三个主要苗头化合物
YS1(最优命中):
• LC/MS 蛋白标记:在 1 μM / 2 h 条件下达到接近 100% 共价标记率 • IC₅₀(体外 BTK 抑制):30 nM • IC₅₀(细胞水平,Mino 细胞):107 nM(BTK 自磷酸化抑制) • 选择性:激酶组学(362 个激酶,300 nM)仅 6 个激酶抑制 > 40%(3 个为 TK 组家族成员:JAK3 85.1%、BMX 95.3%、TEC 45.6%) • 蛋白质组学选择性:Probe 4 竞争实验中,全细胞蛋白质组(3416 个蛋白)中仅 TEC 激酶显著被竞争 • kinact/KI = 915.85 M⁻¹s⁻¹;kinact = 0.004887 s⁻¹;KI = 5.34 μM
YS2:
• IC₅₀(体外):77 μM • 分辨率 1.27 Å 晶体结构(PDB: 9ZLM),RMSD = 0.41 Å • 延伸至 BTK "背口袋"(back pocket),与 Gatekeeper 和 C-helix 之间的空间相互作用
YS3:
• IC₅₀(体外):8.4 μM • AF3 预测与晶体结构存在偏差(约 1 个键旋转的差异),推测与多构象结合模式有关 • 与 CC-292 的结合口袋相似,后者也已知具有多构象
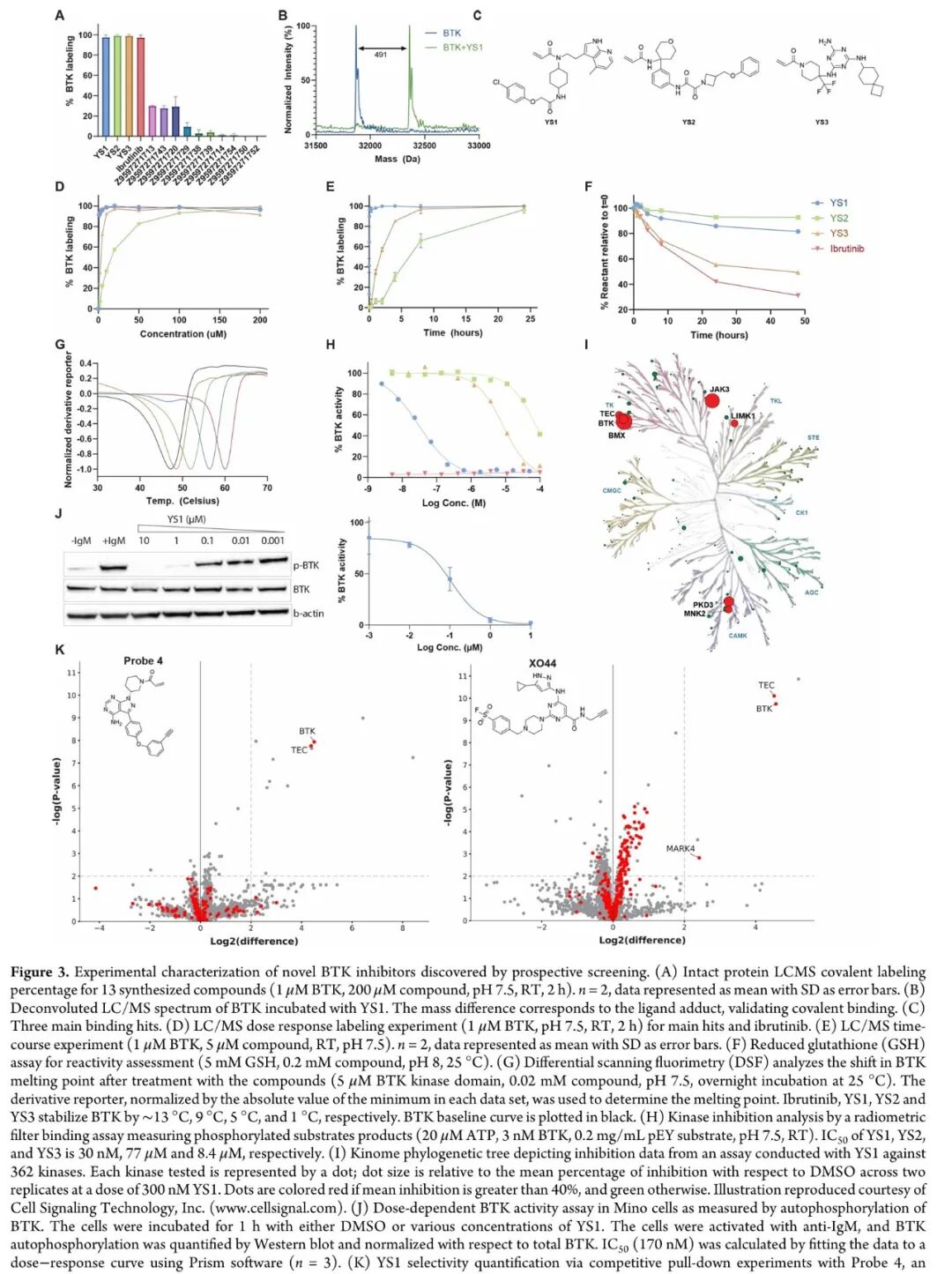
5.3 YS1 的结合模式:全新口袋发现
晶体学揭示了 YS1 与 BTK 结合的独特模式:
YS1 并非传统的 front-pocket 或 back-pocket 结合剂,而是从 P-loop(甘氨酸富集环)下方伸入,将其氯代苯基延伸至由以下残基围成的全新亚口袋:
• M437(靠近 N-lobe 的 C-helix 附近) • L542 / Y545 / V546(激活环 N 端)
这一结合模式伴随:
• P-loop 向开放构象重排(accommodating the ligand) • F413 侧链采取异常朝向(指向 ATP 结合口袋而非激活环) • Y551 未进入"被隔离状态(sequestered state)",与 front-pocket 抑制剂不同
意义:YS1 的发现拓展了我们对 BTK 可成药口袋的认知,揭示了一个此前未被利用的新亚口袋,为克服 BTK 抑制剂耐药性提供了新的结构基础。
5.4 晶体结构验证 AF3 预测精度
| 0.50 Å | |||
| 0.41 Å | |||
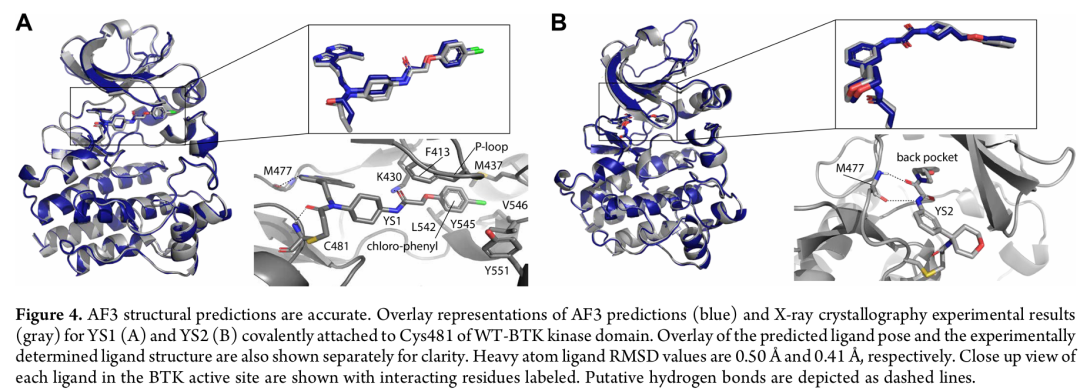
YS1 AF3 预测还正确判断了环己基的反式(trans)异构体构型。
六、深度讨论
6.1 AF3 何以优于传统对接?
结构原因分析:
1. 共价键位点指定:明确指定共价键位点可将 AF3 的结合口袋预测准确率从 81% 提升至 93%(原 AF3 论文数据) 2. 蛋白骨架柔性:AF3 可采样蛋白骨架构象空间,处理 induced-fit 效应。相比之下,传统对接使用固定骨架——当 DOCKovalent 用相同库对 BTK 进行静态结构对接时,YS1 仅排在第 15 百分位(AF3 mPAE 方法排在前 0.05%),且对接构象与晶体结构不符(F413 侧链产生碰撞) 3. 隐蔽口袋采样:AF3 暴露了 YS1 结合的 P-loop 下方新亚口袋 4. E2E 学习的隐性物理知识:Rosetta 重打分 AF3 模型即可优于传统对接,表明 AF3 生成的结构具有足够的物理合理性
6.2 mPAE 的本质与局限
为何 mPAE 有效但 mPAE 与亲和力无关?
这是本文一个深刻且反直觉的发现:mPAE 与结合亲和力(如 IC₅₀、Kd)无显著相关性(Pearson r = 无显著相关),但它能有效区分活性/非活性分子。
作者的解释:mPAE 是一个"粗粒度(coarse-grained)"的分类指标,本质上反映的是配体是否能在预测结合口袋中形成稳定的、几何合理的结合模式,而非结合自由能的精确量化。这与 Masters et al.(2025)的研究结论一致:AF3 等共折叠模型并不依赖精确的物理相互作用来放置配体,而是基于训练集中的统计模式。
选择性预测的局限:
交叉对接实验(cross-docking)显示,对于位点相近的激酶(如 BTK、BMX、ITK 等均含有 Cys 在同源位置),mPAE 无法有效区分。因此 mPAE 目前不适用于激酶选择性预测。
6.3 实用性的瓶颈与解决方案
速度瓶颈:每个配体约需 266 秒(不含蛋白 MSA 生成的 ~22 分钟),对数十亿量级的超大型虚拟库筛选不可行。
建议的级联筛选策略:
超大型虚拟库(10⁹ 量级)
↓ 快速传统对接(毫秒/分子)
前 10⁶ 候选
↓ AF3 + mPAE 精筛(~266 s/分子)
最终候选(~0.05%)
↓ 实验合成与验证弹头多样性限制:本研究仅针对丙烯酰胺弹头(靶向 Cys),无法处理多弹头库中不同亲电试剂反应活性差异的问题。扩展至其他弹头类型(靶向 Ser、Lys、Tyr、His)和其他亲核残基是重要的未来方向。
6.4 AF3 的物理局限性
若干研究表明 AF3 存在物理建模的缺陷:
• YS3 的错误预测:将疏水基团朝向溶剂,可能反映 AF3 对疏水效应(hydrophobic effect)建模不足(与 Childs et al. 2025 一致) • 共价键几何畸变:AF3 共价共折叠倾向于产生键长/键角几何不合理的共价键,但整体配体姿态仍然准确(结构"补偿") • 不依赖精确物理相互作用:Masters et al.(2025)的对抗性实验(adversarial experiments)表明,AF3 对结合位点局部突变不敏感
这些局限性提示:将 AF3 的统计学习与物理化学知识(如 Rosetta 能量函数、力场)结合,是未来改进共折叠方法的重要方向。
七、与相关工作的比较
八、方法学要点(对实践者的启示)
8.1 如何复现/应用本方法
1. 蛋白输入:仅需氨基酸序列(无需晶体结构),AF3 服务器免费可用(alphafoldserver.com) 2. 配体输入:SMILES → 三维构象生成(如 RDKit、OMEGA)→ 指定共价键原子对 3. 关键参数: • mPAE 阈值建议:< 0.9 Å(约 0.05% 命中率,可按需调整) • 相似性过滤:Tc < 0.35(Morgan 指纹)排除与已知抑制剂相似的化合物 4. 计算资源:MSA 生成约需 22 min/蛋白(20 CPU),AF3 推理约 266 s/配体(GPU,型号依赖)
8.2 注意事项
• 对于已有大量已知抑制剂的靶点(如 BTK),需严格进行新颖性过滤(novelty filter),避免重发现已知骨架 • mPAE 在同源位点的激酶之间分辨率有限,选择性评估仍需体外实验 • YS3 案例提醒:对 AF3 预测构象存疑时,应进行晶体学验证,不能完全信赖预测结合模式 • 对于共价键几何(键长/键角),AF3 预测可能偏差较大,需结合化学合理性判断
九、总结与展望
核心结论
本文通过以下五点系统性地证明 AF3 可以实际用于共价药物发现:
1. ✅ 构建了 COValid,填补了共价 VS 基准数据集的空白 2. ✅ 证明 AF3 mPAE 指标在所有测试靶点均大幅优于传统共价对接工具 3. ✅ 证明其优越性不依赖训练数据泄露(消融实验) 4. ✅ 在非共价 VS(DUDE-Z)上同样优于传统方法 5. ✅ 前瞻性发现了全新骨架的 BTK 共价抑制剂 YS1,并通过晶体学验证了 AF3 预测的亚埃精度
对领域的影响
• 方法论层面:为 AI 辅助共价药物发现提供了经过充分验证的工作流程 • 药物发现层面:开启了利用蛋白构象柔性进行大规模虚拟筛选的新时代,对传统对接难以处理的柔性靶点尤为重要 • 生物学层面:YS1 揭示了 BTK 的全新结合亚口袋,为克服 BTK 抑制剂耐药性提供新思路
未来方向
• 扩展至非激酶靶点的系统性验证 • 开发针对 Ser、Lys、Tyr、His 等其他亲核残基的弹头,配合 AF3 共折叠 • 融合物理化学能量函数改善 AF3 对疏水效应的建模 • 建立 AF3 + 快速对接的级联筛选流程,适配十亿量级超大型虚拟库 • 探索 AF3 建模共价反应中间态(prereaction state)的可行性,以更好捕捉 kinact 相关信息
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢