
UniProt 数据库中已收录超过 2.5 亿条蛋白质序列,但其中拥有实验验证功能注释的不到 0.1%。这意味着,绝大多数已知蛋白质的功能仍然是未知的。传统的同源转移方法只在序列相似性足够高时才可靠,而深度学习方法虽然强大,却大多将 Gene Ontology(GO)术语视为独立的分类目标,无法捕捉蛋白质功能预测所需的多模态整合推理——而这恰恰是生物学家注释未知蛋白质时真正在做的事情:综合序列、结构、结构域、进化上下文和相互作用网络,构建一个连贯的功能假说。
近日,Arc Institute、多伦多大学、Vector Institute 和 University Health Network 的联合团队在 bioRxiv 上发布了 BioReason-Pro,这是首个用于蛋白质功能预测的多模态推理大语言模型。它不仅在 GO 术语预测上达到了 73.6% 的 F_max,更在 27 位蛋白质专家的盲评中取得了惊人的成绩——在 79% 的评估案例中,专家认为 BioReason-Pro 的注释质量不低于甚至优于 UniProt 的人工审编条目。模型权重、代码、数据集和在线推理平台全部开源。

GO-GPT:将 Gene Ontology 预测转化为序列生成任务
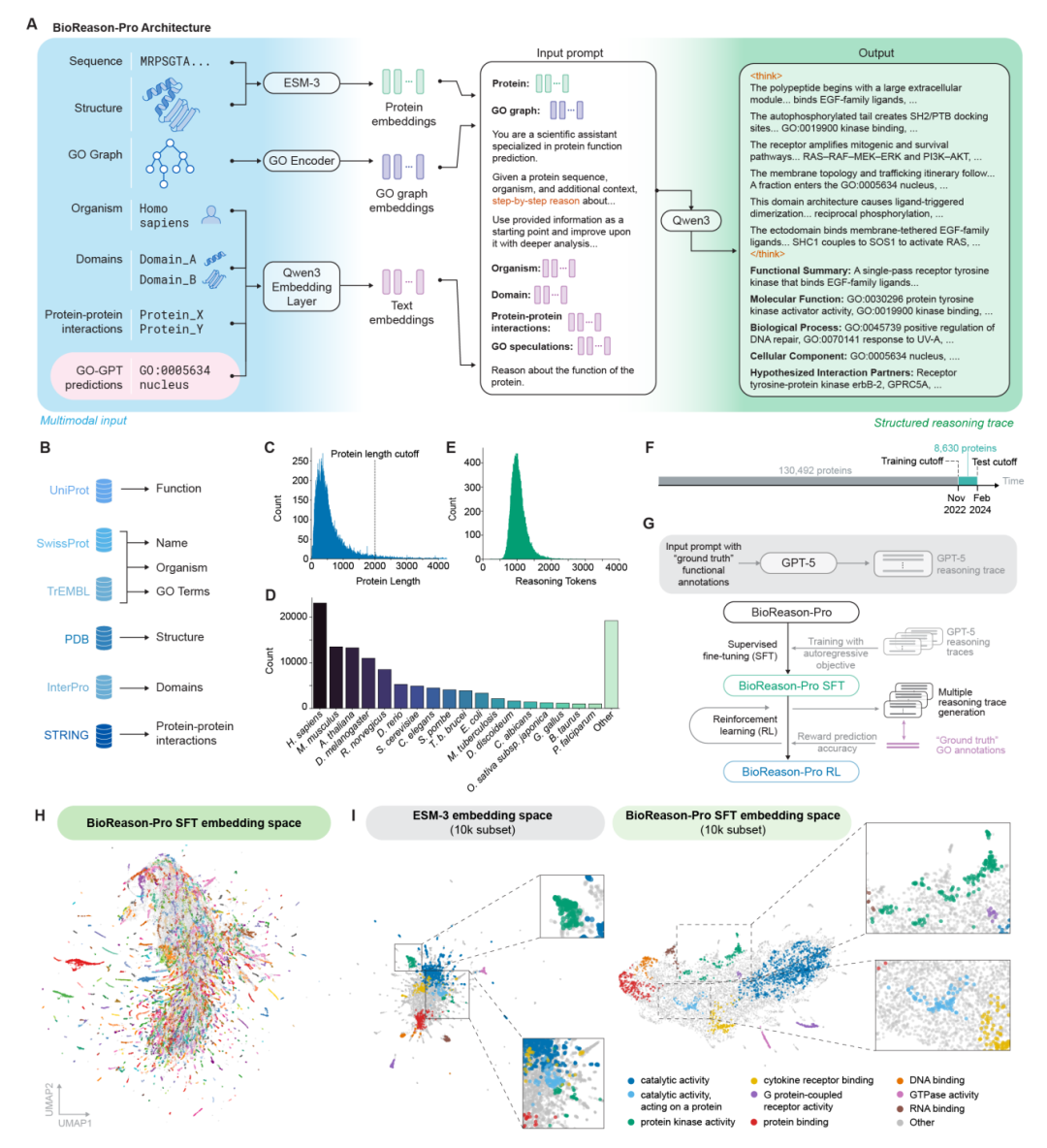
BioReason-Pro 的一个重要创新是其上游组件 GO-GPT——一个自回归 Transformer,将 GO 术语预测重新定义为一个序列生成任务。
现有的 GO 预测方法几乎都将每个术语视为独立的分类目标。但 Gene Ontology 本身是一个有向无环图(DAG),从通用的根概念到高度特异的叶节点层层递进,且三个方面(分子功能 MF、生物过程 BP、细胞组分 CC)之间存在跨方面的依赖关系。GO-GPT 通过自回归生成的方式天然地捕捉了这些层级关系和跨方面依赖:每个 GO 术语的预测都以蛋白质嵌入(来自 ESM2)、目标物种和所有此前已生成的术语为条件。
在 CAFA 5 框架的时间切分测试集上,GO-GPT 以加权 F_max 0.65(贪心解码)超越了 CAFA 5 竞赛中排名最高的公开方法 InterLabelGO+(0.63)。通过对每个蛋白质采样 10 条独立轨迹并以术语出现频率估计概率,性能进一步提升至 0.67;选取 10 条中的最佳样本则可达 0.70,显示出解码策略仍有可观的提升空间。
GO-GPT 的学习表征同样值得关注。模型为每个物种学习了独立的嵌入向量,对这些嵌入计算余弦相似性并构建系统树后,所得到的树与已知的系统发育关系高度一致(Mantel 检验 p = 4 × 10⁻³),说明模型在没有任何显式监督的情况下捕捉到了物种间的进化约束。在 DNA 结合蛋白的注意力分析中,GO-GPT 预测 DNA 结合功能(GO:0003677)时的残基级注意力与已知的 DNA 结合位点高度富集(平均 AUROC = 0.81 ± 0.06,top-20% 富集倍数 = 2.8x),表明模型确实从序列特征中学到了功能相关信号。
从 SFT 到 RL:教 LLM 像生物学家一样推理
BioReason-Pro 构建在 Qwen3-4B 基础之上,深度整合了 ESM3 的残基级蛋白质嵌入、一个 GO 图编码器,以及包括目标物种、InterPro 结构域注释、STRING 蛋白-蛋白相互作用和 GO-GPT 预测结果在内的生物学上下文。模型输入这些多模态信息后,以链式推理(chain-of-thought)的方式生成结构化推理轨迹,从结构域分析出发,逐步推导出功能假说。
训练蛋白质功能推理模型的一个核心难题是缺乏大规模的人类撰写推理轨迹。团队采用了一个务实的方案:使用 GPT-5 为 133,492 个蛋白质(覆盖 3,135 个物种)生成合成推理轨迹,然后对 Qwen3-4B 进行 SFT 训练。这教会了模型如何生成生物学推理链。随后,团队使用 GSPO(Group Sequence Policy Optimization)进行强化学习,以预测 GO 术语与真实标签之间的加权 F_max 作为奖励信号,直接优化 GO 术语预测的准确性。
RL 的效果颇为有趣。训练过程中,奖励信号稳步上升的同时推理轨迹反而变短了,说明模型学会了精简冗余推理而非简单地缩短输出。逐蛋白质对比显示,RL 版本的推理轨迹平均比 SFT 版本短 60 个词(Wilcoxon p < 10⁻³⁰⁰),但在较短轨迹长度范围内 RL 的优势更加显著——这意味着强化学习产生了更高效的推理。
自动化评测与人类专家盲评
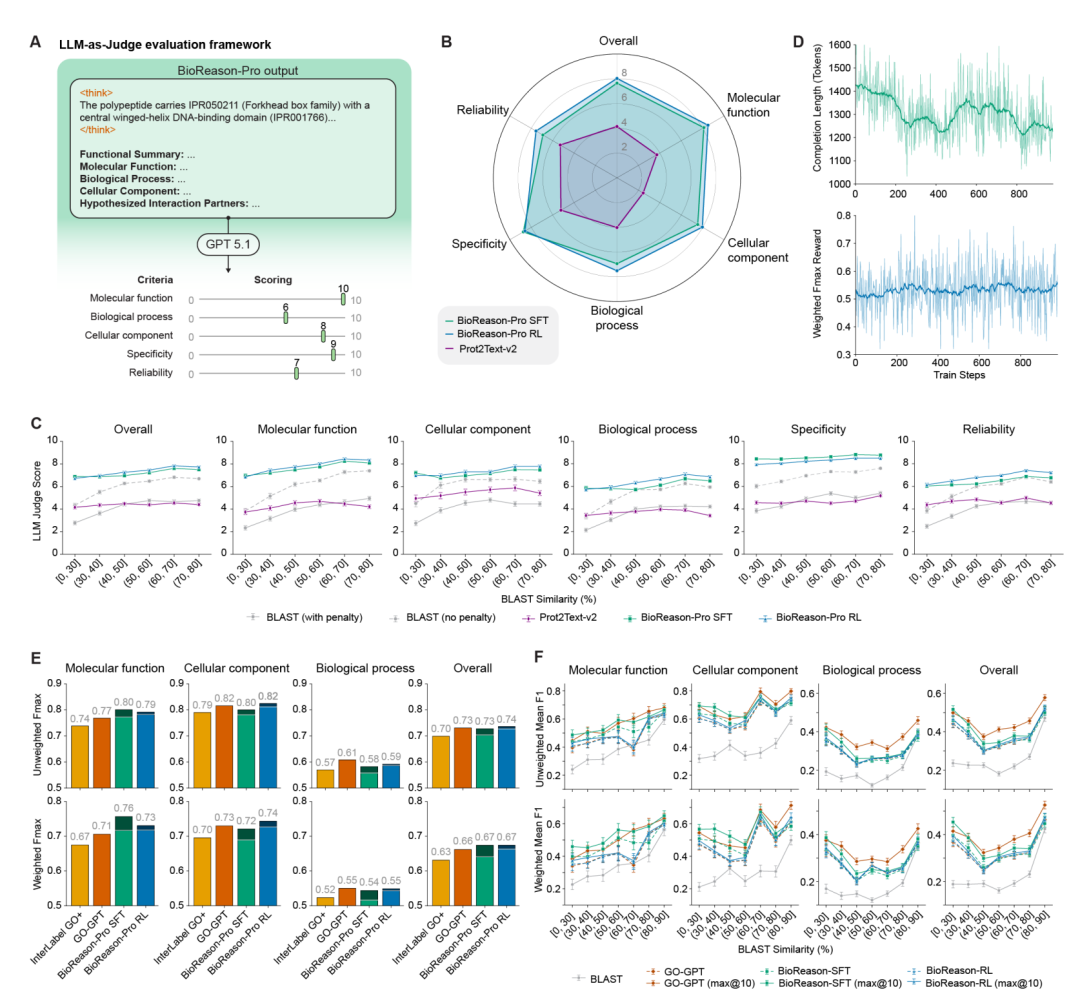
BioReason-Pro 的评测体系值得细说,因为蛋白质功能预测的评测本身就是一个棘手问题——标准的文本相似度指标(ROUGE、BERTScore)衡量的是表面重叠而非生物学正确性。
团队使用 GPT-5.1 作为自动化评委,在分子功能、生物过程、细胞组分、特异性和可靠性五个维度上对模型预测打分。BioReason-Pro RL 的平均得分为 8.03/10,SFT 为 7.65/10,而此前最佳方法 Prot2Text-v2 仅为 4.15/10。尤其值得注意的是,模型性能在不同序列相似性区间上保持稳定——即使测试蛋白与训练集的最佳比对相似度很低,BioReason-Pro 仍维持 7-8.5 分的水平,而 BLAST 基线则随相似性降低急剧衰退。
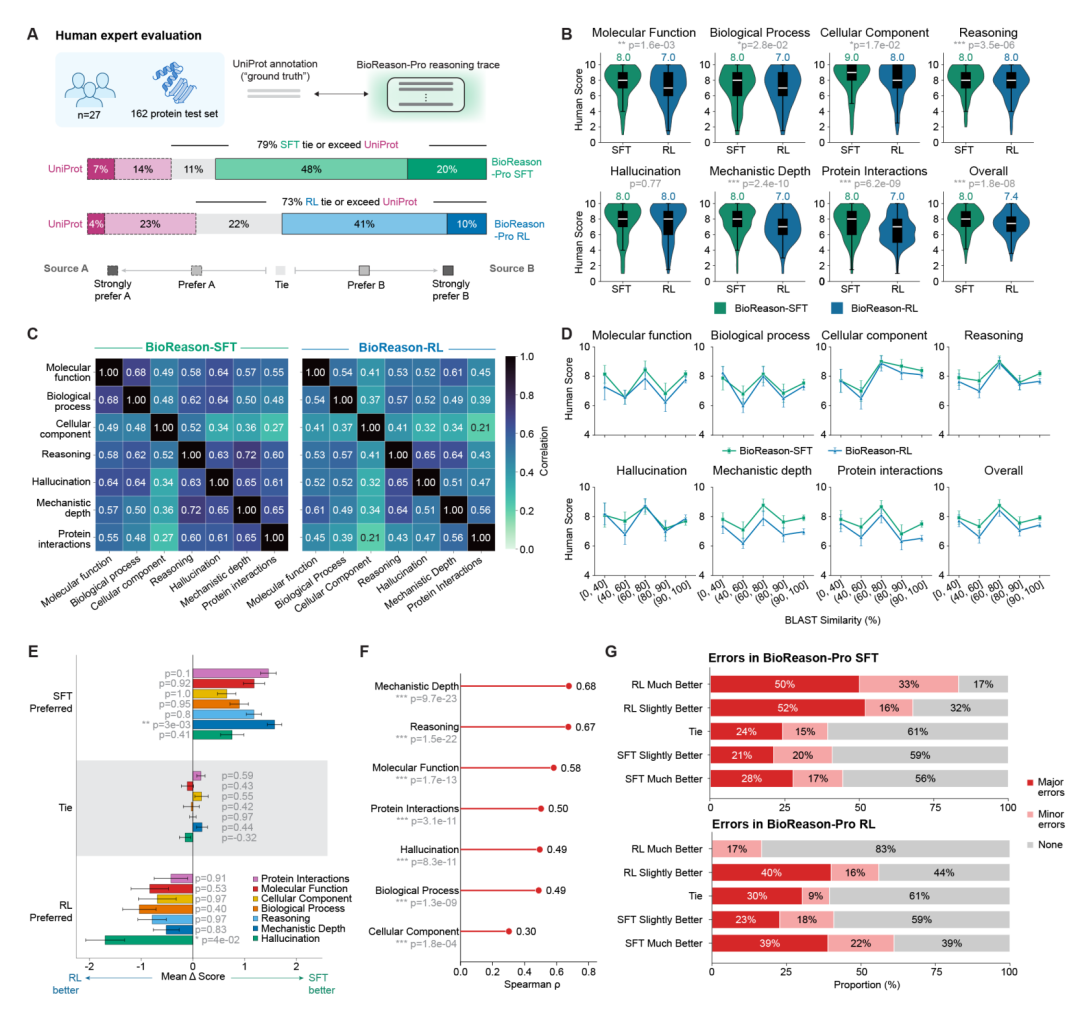
更有说服力的是 27 位蛋白质专家的盲评实验。评审者对模型身份完全不知情,可以查阅外部资源和最新文献。在 162 个随机选取的测试蛋白上,SFT 版本在 79% 的案例中被认为不逊于或优于 UniProt 人工审编条目,RL 版本为 73%。SFT 在机制深度维度上更胜一筹(p = 3 × 10⁻³),而 RL 则在减少幻觉方面表现更优(p = 4 × 10⁻²)——强化学习在提升 GO 术语准确性的同时,也有效压制了捏造的生物学声明,但代价是牺牲了部分机制假说的深度。
亮点案例:从序列到结构界面的全新预测
论文中两个案例研究极具说服力地展示了 BioReason-Pro 的推理深度,远超简单的结构域注释重述。
案例一:eEFSec 与 SBP2 结合伙伴的 de novo 预测。eEFSec(P57772)是硒代半胱氨酸特异性延伸因子,与训练集中最近序列仅有 44% 相同性。BioReason-Pro 从结构域架构出发,识别出 N 端的经典翻译 GTPase 核心和 C 端的硒代半胱氨酸特异性结构域,推断出该蛋白不是通用的 tRNA 递送因子,而是仅识别硒代半胱氨酰-tRNA(Sec)。在没有提供任何蛋白-蛋白相互作用信息的条件下,SFT 版本从架构约束出发,推理出 C 端 RIFT 结构域暗示存在一个桥接 SECIS 元件与核糖体解码位点的护送因子,并直接命名了 SBP2(SECIS 结合蛋白 2)。这一预测已被共免疫沉淀和 2.8 Å 冷冻电镜硒体结构(PDB 7ZJW)实验验证。更引人注目的是,从预测伙伴名称前一个 token 提取的残基级注意力精确定位到 RIFT 结构域表面,与冷冻电镜解析的 SECIS RNA 结合界面高度重合(Mann-Whitney U 检验,p = 2.3 × 10⁻⁵)。作为对比,GPT-5.2 Thinking High 将该蛋白错误地鉴定为 septin 家族 GTPase。
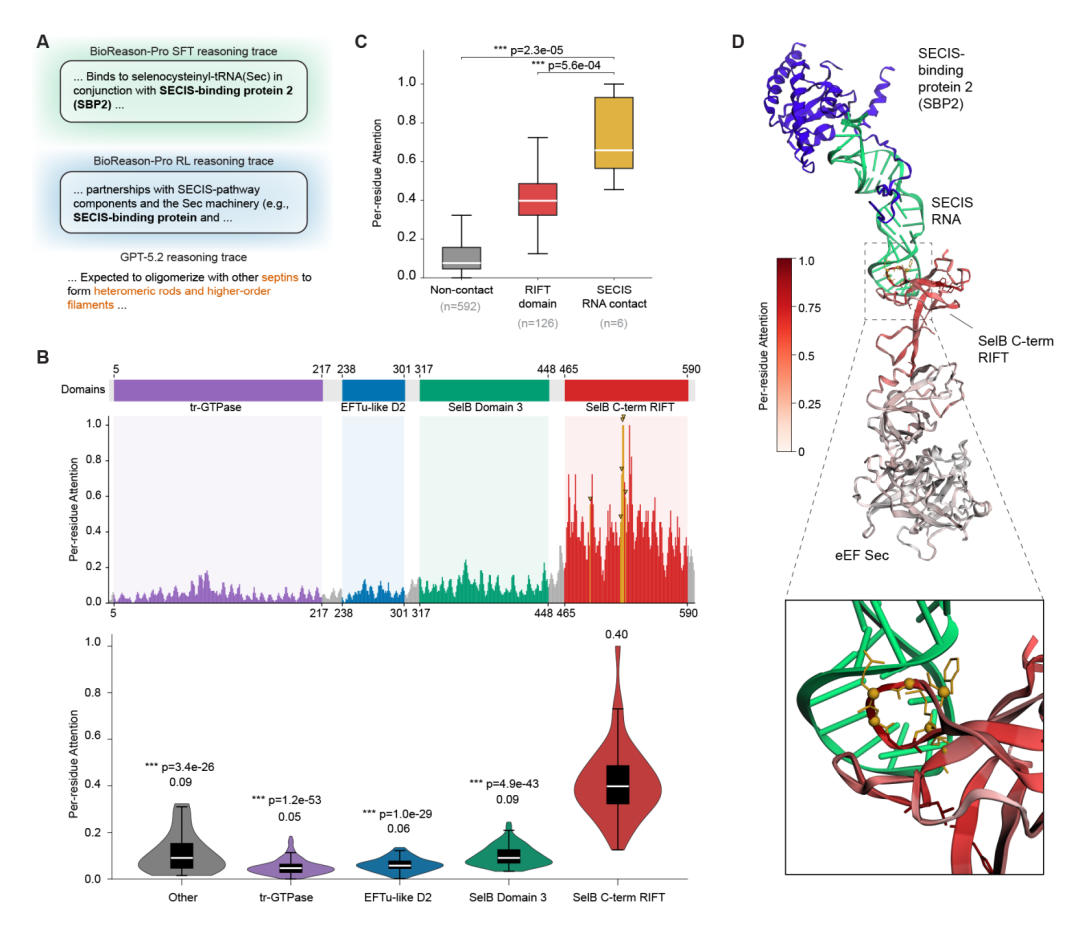
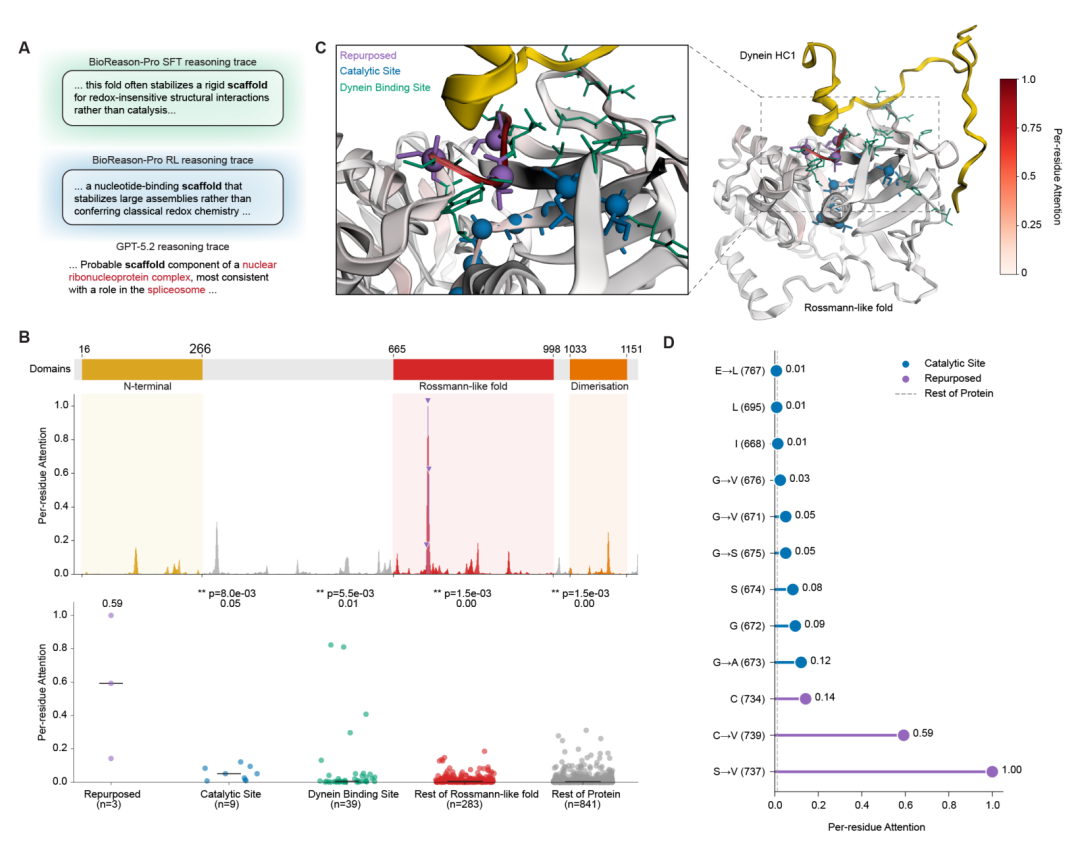
案例二:CFAP61 的结构推理超越结构域注释。CFAP61(Q8NHU2)与训练集最近序列仅 29% 相同性,其结构域包含一个 Rossmann 样 FAD/NAD(P) 结合超家族折叠,这在标准数据库查询中会被错误归类为氧化还原酶。事实上,CFAP61 是纤毛轴丝中一个非酶促支架蛋白。BioReason-Pro 没有孤立地解读该结构域,而是综合考虑了其两侧的 N 端轴丝靶向模块和 C 端二聚化结构域,以及蛋白质所属的纤毛特异性家族,推断出 Rossmann 样结构域提供的是稳定的结构核心而非催化功能。残基级注意力分析进一步揭示,模型最关注的恰恰是 Rossmann 结构域中三个已退化的催化位点残基(C734, V737, V739),而这三个残基在冷冻电镜轴丝结构(PDB 8J07)中正是与 dynein 重链 1 形成直接接触的界面。模型在单残基分辨率上学到了催化位点向蛋白-蛋白相互作用界面的进化转用,这是仅靠同源转移或结构域查询无法实现的。
边界探索与局限性
文章也坦诚讨论了 BioReason-Pro 的能力边界。
在短肽(< 50 个氨基酸)上,模型性能系统性下降。SFT 版本在缺乏可识别结构域的短肽上会捏造 InterPro 条目——例如为一个 GAD65 表位指定了肌球蛋白家族结构域,并围绕虚构结构域编织出看似合理的机制叙事。RL 版本则从不捏造结构域,而是明确承认缺乏可识别特征,但功能预测的特异性也随之降低。
对 AI 生成的蛋白质(如 Evo 1.5 设计的两个 anti-CRISPR 蛋白),模型对物种标签高度敏感——同一条 EvoAcr1 序列在不同物种标签下分别被预测为核糖体蛋白、DNA 结合转录抑制因子、宿主-病原体膜效应物和翻译起始因子。这一现象的生物学基础在于不同物种的蛋白质组差异确实会塑造功能偏好,但也暴露了模型对上下文信号的过度依赖。
此外,模型的训练数据以 2022 年 11 月为截止,无法捕捉此后新发现的功能。例如,BRINP2 被准确识别为核转录抑制因子(与训练期间的注释一致),但其最近才发现的作为 BRP 前体肽的激素原角色则未被预测到。
全面开源与在线平台
BioReason-Pro 的开源力度在同类工作中极为突出。全部模型权重(GO-GPT、BioReason-Pro SFT 和 RL 三个版本)、训练代码和策展数据集均通过 HuggingFace 发布。团队还搭建了在线推理平台 bioreason.net,提供即时蛋白质功能推理,并在 bioreason.net/atlas 上公开了超过 223,000 个蛋白质(包括 Human Protein Atlas 全覆盖)的预计算预测结果。
思考
BioReason-Pro 代表了蛋白质功能预测范式的一次重要转变:从将 GO 术语视为孤立分类标签,走向像生物学家一样进行多模态整合推理。它将蛋白质基础模型(ESM3)的表征深度与 LLM 的链式推理能力结合在一起,实现了从结构域分析到功能假说的完整推理链。两个案例研究表明,这种推理不仅仅是重新表述已有注释——模型能够在单残基分辨率上进行结构推理,其内部表征与实验解析的结构界面精确对应。
但这也引发了更深层的问题:当一个 4B 参数的 LLM 就能在 79% 的案例中匹敌人类专家审编,蛋白质功能注释的 gold standard 本身是否需要被重新定义?尤其是考虑到 UniProt 的人工审编覆盖率极低、更新周期漫长,BioReason-Pro 这类系统的大规模部署或许将从根本上改变计算生物学的注释基础设施。
MindDance 此前在 AI工具正在接管生命科学实验室?中就指出,将 LLM 推理能力与领域专用编码器结合是 AI for Science 的核心方向之一。BioReason-Pro 为这一方向提供了迄今最有力的实证。
论文链接:
https://www.biorxiv.org/content/10.64898/2026.03.19.712954v1
GitHub:
https://github.com/bowang-lab/BioReason-Pro
在线平台:
https://bioreason.net/
模型权重:
https://huggingface.co/collections/wanglab/bioreason-pro
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢