
蛋白质与小分子配体之间的识别与结合,是药物发挥作用的分子基础。在这一过程中,氢键、卤键、盐桥、阳离子–π相互作用以及π–π相互作用等多种非共价相互作用(Noncovalent Interactions, NCIs)往往并非孤立存在,而是相互影响、彼此调控,形成复杂的“分子间作用力网络”。这些相互作用之间究竟是共同增强的协同效应,还是相互减弱的拮抗关系?这一问题对于药物分子的理性设计具有重要意义。
近日,中国科学院上海药物研究所朱维良、华东理工大学徐志建团队基于数据库统计分析,并结合量子化学/分子力学计算方法系统研究了蛋白–配体体系中多种非共价相互作用之间的协同与拮抗规律。相关成果以题为“Insights into Protein–Ligand Noncovalent Interaction Networks: A Database Survey and Quantum Chemistry Calculation Study”1的封面文章发表于 Journal of Medicinal Chemistry。

工作介绍
研究人员首先从Protein Data Bank(PDB)数据库中提取结构数据,按照筛选流程保留分辨率优于3 Å的蛋白–配体复合物共109,344个。在去除辅因子及其他明显非药物相关小分子后,最终获得139,489个高质量配体用于后续分析。
针对配体中可能参与氢键、卤键、阳离子–π、π–π相互作用及盐桥的供体与受体功能基团进行系统统计。结果显示,仅有15.85%的配体只包含单一类型的非共价相互作用(NCI)基团,绝大多数配体(71.55%)同时包含三种及以上类型的NCI基团(图1)。这一结果表明,在先导化合物优化过程中,可以通过合理引入更多类型的NCI基团来提升其与靶标的结合潜力。
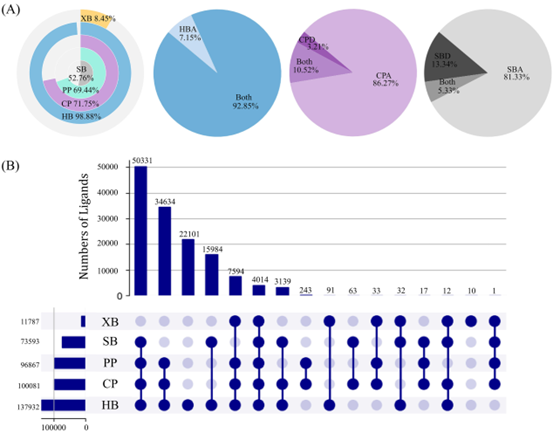
图1. 配体中NCI基团的分布情况 (A)含有不同类型NCI基团的配体所占比例,其中氢键(HB)、阳离子–π相互作用(CP)和盐桥(SB)进一步根据供体、受体及同时具有供体与受体进行分类(饼图表示);(B)UpSet图展示了同时包含一种至五种NCI基团的配体数量分布情况。
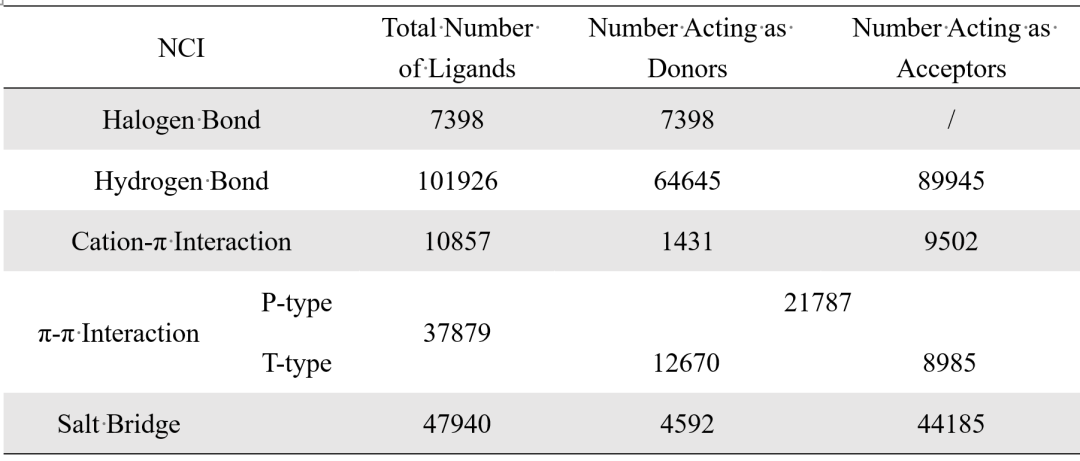
在此基础上,研究进一步依据几何判据筛选并统计配体与蛋白之间实际形成的非共价相互作用情况(表1)。结果表明,配体所含的氢键及卤键基团与靶标形成实际相互作用的概率均超过50%,其中氢键受体与供体的实际形成比例分别为65.21%和50.48%,卤键供体占比62.76%,显示出较高的成键倾向。而T型π–π相互作用供体和阳离子–π相互作用受体基团与靶标实际作用比例较低,表明此类相互作用在蛋白–配体体系中对几何构型和化学环境具有更高要求。
表1. 几何筛选条件的配体数量
在统计分析的基础上,研究人员选取具有代表性的体系进行QM/MM结构优化,并计算协同与拮抗能(ΔEsyn)(图2)。以0.41 kcal/mol(对应约两倍活性差异的能量尺度)作为显著协同或拮抗效应的判据进行分析。结果显示,CPA–SBA(1.31±0.78 kcal/mol)、CPD–SBD(0.41±0.21 kcal/mol)、HBA–SBA(0.84±0.80 kcal/mol)及SBD–PPTA(0.58±0.72 kcal/mol)是典型的强拮抗效应组合,其中CPA–SBA表现最为显著。而HBD–HBA组合(-0.73±1.08 kcal/mol)表现出最强的协同效应。整体而言,在蛋白–配体体系中,多数非共价相互作用之间近似呈现加和关系,仅少数特定NCI组合会显著改变整体相互作用能量。
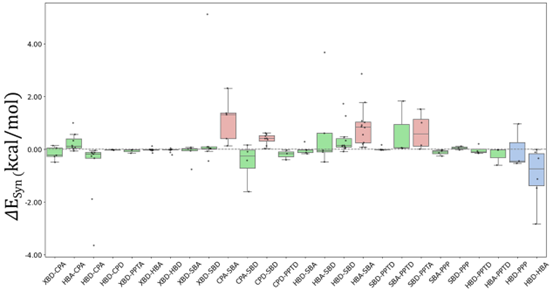
图2. 两种 NCI体系中非共价相互作用组合的协同与拮抗能量分布。
进一步分析发现,当两种NCI位点之间的距离大于10 Å时,协同或拮抗效应整体较弱;当距离小于7.5 Å时,协同与拮抗效应增强,尤其拮抗效应更为突出(图3B)。这说明简单增加相互作用数量并不必然增强结合亲和力,反而可能引发不利的相互作用,从而产生“1 + 1 < 2”效应。在三种NCI体系中,总协同/拮抗能可较好地近似为相应两种NCI体系协同/拮抗能之和(R²=0.980)(图3C)。这一结果进一步强调,合理构建相互作用网络结构比单纯增加功能基团数量更为关键。此外,研究选取了六个具有代表性的强协同或拮抗体系进行了电子层面的分析(图4)。
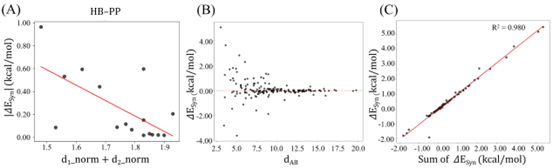
图3. 多种NCI体系中的相关性 (A)HB–PP体系中d1_norm + d2_norm与两种NCI体系中|ΔESyn|之间的相关性;(B)两种NCI体系中相互作用残基位点距离与ΔESyn之间的相关性;(C)三种NCI 体系ΔESyn与对应两种NCI能量之和之间的相关性。
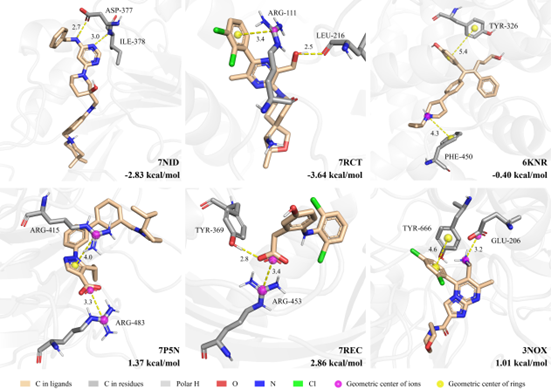
图4. 蛋白质–配体体系中具有代表性的协同(上)和拮抗(下)相互作用。
该研究从大规模数据统计与量子化学计算两个层面系统揭示了蛋白–配体体系中复杂非共价相互作用网络的协同与拮抗规律,为活性化合物的结构优化提供了理论指导。
该论文第一作者为中国科学院上海药物研究所硕士研究生罗睿童,通讯作者为中国科学院上海药物研究所的朱维良研究员以及华东理工大学药学院特聘教授徐志建。该项研究工作得到了国家自然科学基金和科技部重点研发项目的资助。徐志建/朱维良团队多年来致力于卤键领域的相关研究,首次阐明了卤键作用的本质2,揭示了PDB数据库中卤键等分子间作用力被普遍低估的现象3,研究了生物体系中存在的氢键诱导的氟卤键4。
参考文献
[1]Insights into Protein–Ligand Noncovalent Interaction Networks: A Database Survey and Quantum Chemistry Calculation Study. J. Med. Chem. 2026, 69 (6), 6965–6975
https://doi.org/10.1021/acs.jmedchem.5c03216
[2] Halogen bonding in differently charged complexes: basic profile, essential interaction terms and intrinsic σ-hole. Phys. Chem. Chem. Phys. 2019, 21 (27), 15106– 15119
[3] The Underestimated Halogen Bonds Forming with Protein Side Chains in Drug Discovery and Design. J. Chem. Inf. Model. 2017, 57 (1), 22−26.
[4] Hydrogen Bond-Induced Binding between Organofluorine and Protein via Fluorine Atoms: A Database Survey and Quantum Chemistry Calculation Study. J. Med. Chem. 2025, 68 (14): 14882-14894.
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢