

文献来源:Fajar A.T.N. et al., Advanced Science, 2026, DOI: 10.1002/advs.202523042
发表机构:日本九州大学
引言
如果有一个 AI,不需要人类告诉它"这个分子可能好用",就能自己从零设计出有效的新材料分子——你会怎么想?
这不是科幻。2026年3月,日本九州大学的研究团队在《Advanced Science》上发表了一项令人振奋的研究:他们用生成式 AI,首次自动设计出了用于钙钛矿太阳能电池的钝化分子,并在实验室完成了验证。
为什么这件事很重要?
钙钛矿太阳能电池(PSC)近年来发展迅猛,单结效率已突破 27%,被认为是下一代光伏技术的明星选手。但它有一个卡脖子的问题:界面缺陷。
缺陷会导致载流子复合,拉低效率、降低寿命。科学家的解决思路是:在钙钛矿表面涂上一层"钝化分子",把缺陷堵住。听起来简单,但现实是——好用的钝化分子要靠人类专家凭经验一个个试,效率极低,能搜索的化学空间也极其有限。
化学空间据估计超过 10⁶⁰ 种可合成小分子。用人力逐一筛选?那要试到宇宙终结。
生成式 AI 的范式转变
生成式 AI 提供了从"筛选已知分子"到逆向分子设计的范式转变。合成可及的小分子化学空间估计超过 10⁶⁰,生成模型理论上可在此空间中自由探索。SyntheMol(抗生素发现)和 MatterGen(晶体结构生成)已在各自领域展示了生成式 AI 的威力,但其在 PSC 钝化分子领域的应用至今仍是空白。
这项研究做了什么?
2.1 整体工作流程
研究采用三阶段闭环流水线:
[阶段一] 数据库构建与判别模型训练
文献挖掘 → Data T0(314分子)→ SMILES-X 分类模型
↓
[阶段二] 化学空间扩展与生成
数据增强(PubChem相似性检索)→ Data T1(>11,000分子)
→ GPT-2 微调(3轮迭代)→ 生成 >100,000 新分子
→ 7维物化性质过滤 → ~8,000 候选分子
→ 聚类分析(10簇)→ 10 代表性分子
↓
[阶段三] 实验验证
3 个分子 → 反型 PSC 器件制备 → 光电性能表征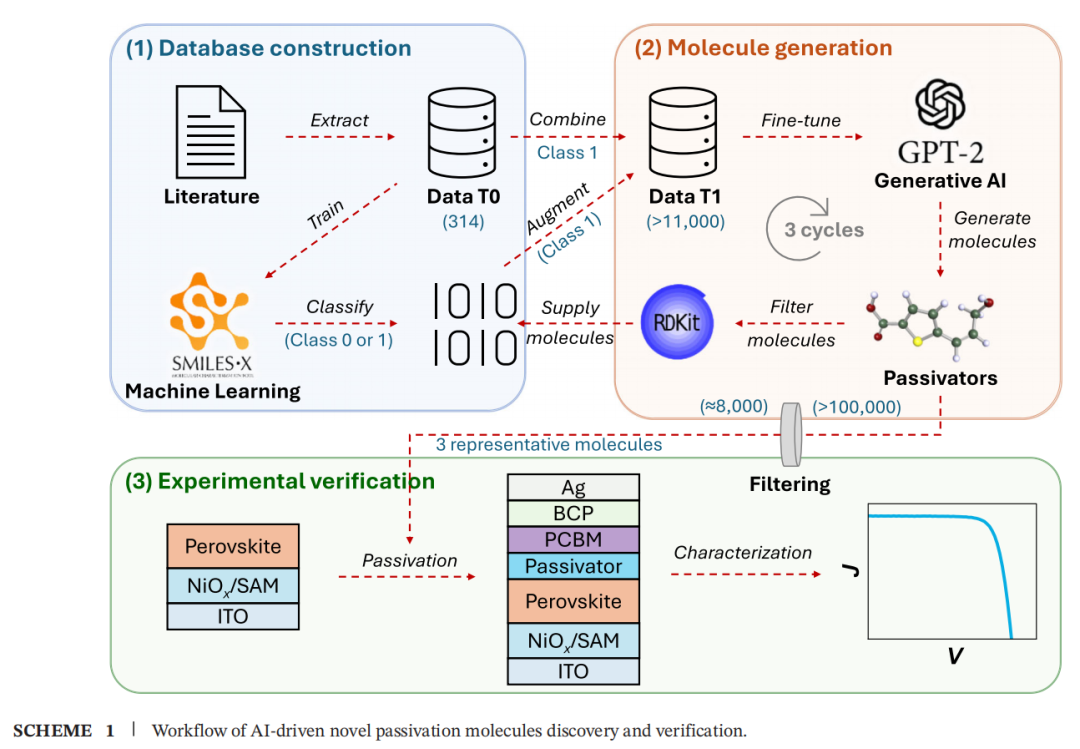
2.2 数据集构建(Data T0)
数据来源:系统性挖掘综述文章及相关一手文献,手动核实原始出版物。
数据预处理:
• 去除重复结构,转化为规范 SMILES • 提取初始/最终 PCE 值,计算归一化 PCE 提升量(ΔPCEnorm) • 最终获得 314 个唯一标注分子
分类规则:
• Class 1(有效):ΔPCEnorm ≥ 0.10 • Class 0(无效):ΔPCEnorm < 0.10
数据集特征分析:
• 原子类型:以 C、O、N 为主,含 F、S、Cl 等卤素 • 分子大小:多数小于 20 个原子(最大达 60 原子) • 化学空间(PCA/UMAP):有效与无效分子在化学空间中高度混叠,表明结构-活性关系极为复杂
2.3 判别模型:SMILES-X 分类器
模型架构:SMILES-X 直接以 SMILES 字符串为输入,无需人工特征提取,端到端映射至属性标签,遵循自然语言处理范式。
训练策略:五折交叉验证
性能指标:
| 0.80 | |
| 0.88 |
混淆矩阵(5折汇总):
SMILES-X 与随机森林(Morgan 指纹)性能相当,但无需显式特征提取,可无缝集成至语言模型生成框架,因此被选为判别组件。
2.4 数据增强(Data T-aug)
由于 Data T0 规模过小(314条),不足以直接微调 LLM,研究采用以下策略扩增:
• 从 PubChem 检索与 Data T0 中 Class 1 高效分子(ΔPCEnorm > 0.16)Tanimoto 相似度 ≥ 80% 的分子 • 获得 15,540 个额外条目(Data T-aug) • 其中约 70% 被 SMILES-X 分类为 Class 1 • 合并 Data T0 与 Data T-aug 中的 Class 1 分子,构成微调数据集 Data T1(>11,000 分子)
2.5 生成模型:GPT-2 迭代微调
模型选择:同时训练了 GPT-2 和 LLaMA-2(70亿参数),对比如下:
迭代微调策略:
• Cycle 1:在 Data T1 上初次微调,生成约 30,000 个化学有效、唯一且新颖(CUN)分子 • Cycle 2:将 Cycle 1 中的 Class 1 分子并入训练集,重新微调后生成量显著提升 • Cycle 3:再次扩充训练集,CUN 分子总量突破 100,000
结果统计:
• 总生成量(Data G-all):>100,000 个 CUN 分子 • 预测有效(Data G-class1):87,750 个(占比 >80%) • 化学多样性:Tanimoto 相似度分析显示大多数生成分子与训练集具有中等相似度,而非简单复制——验证了模型真正在探索新化学空间
终止条件:第三轮后停止迭代,以维持约 10 万分子的可管理规模,并防止已被先前研究记录的"模型崩塌"(model collapse)现象。
2.6 多维物化性质过滤
对 87,750 个 Class 1 预测分子依次施加 7 项过滤准则:
过滤后保留约 8000 个候选分子(<10%)。
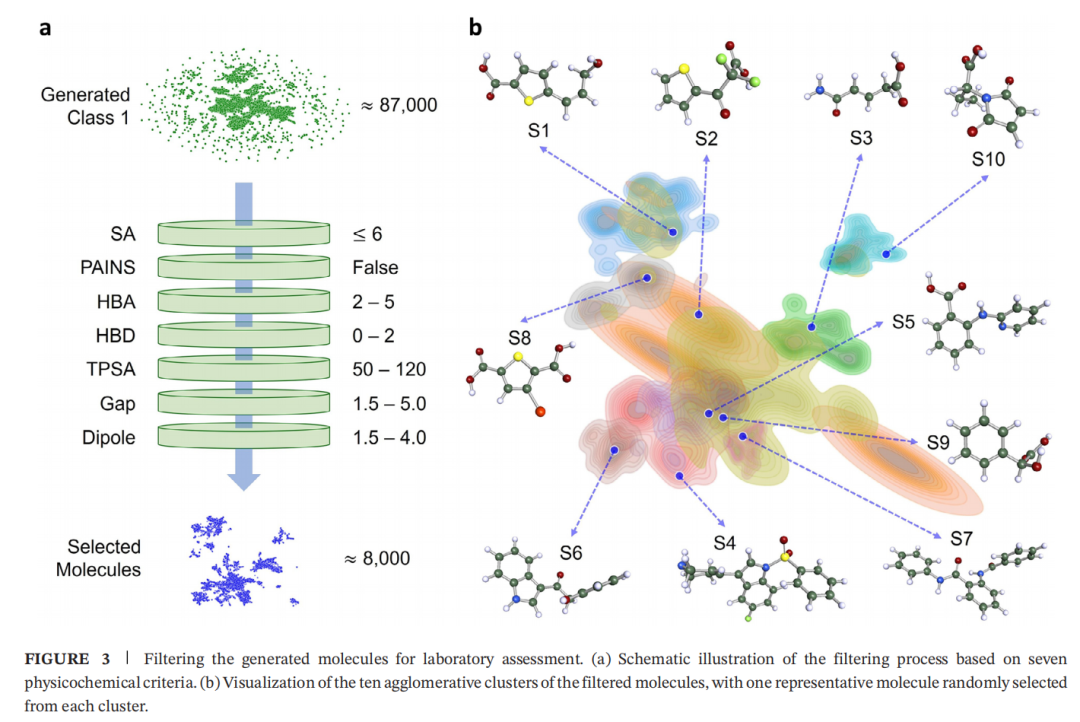
2.7 聚类与代表性分子选取
• 对 8000 个候选分子计算 Morgan 分子指纹 • 采用层次凝聚聚类算法分为 10 个簇 • 每簇随 机选取 1 个代表性分子 • 经领域专家 评估,选出 3 个进入实验验证
最终选出的 3 个分子:
实验结果怎么样?
3 个 AI 推荐的分子,全部在实际器件中展现出钝化效果,开路电压均有提升。其中,4-马来酰亚胺丁酸(MBA)的表现最为亮眼:
开路电压提升:1.08→1.12 V
平均光电转换效率:19.3%→22.2%
迟滞指数大幅下降:0.160→0.036
冠军器件在反向扫描下甚至达到了 24.13% 的效率。研究团队还通过 XPS、光致发光、DFT 计算等多种手段,证实了 MBA 与钙钛矿表面 Pb²⁺ 位点的强配位作用,以及其对界面能级排列的优化效果。
生成式 AI 可以自主提出合成可及、功能有效的钝化分子,为材料加速发现提供了超越传统化学空间探索的全新范式。
写在最后
本研究展示了生成式 AI 在材料发现领域的一个清晰可行的范式:以极小的初始数据集为起点,通过语言模型驱动的迭代生成-筛选-验证闭环,系统性地探索此前人类难以涉足的化学空间。
科研提速的竞赛正在全面展开。从抗生素到电池材料,AI 正在把"试遍所有可能"从不可能变成现实。AI 可以不依赖人类直觉,主动探索未曾被涉足的化学空间,并给出可以直接进实验室验证的候选分子。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢