
DRUGONE
研究人员提出了一种基于血浆蛋白质组学的深度联合学习模型 ProtAIDe-Dx,用于同时诊断与痴呆相关的六种疾病。该模型基于超过17,000名个体的数据进行训练,能够在单次血液检测中输出多疾病的概率性诊断结果。
模型在多个疾病任务中表现出较高的分类性能,平衡准确率达到70%至95%,AUC超过78%。更重要的是,该模型不仅能够区分不同疾病,还能够识别共病情况,并在外部临床队列中与疾病特异性生物标志物表现出一致性。通过模型解释分析,研究人员发现了一系列与神经退行性疾病相关的蛋白网络,这些网络既包含不同疾病共享的生物过程,也包含疾病特异性的分子机制。此外,该模型能够在个体层面识别驱动诊断决策的关键蛋白。总体而言,该研究表明,基于血浆蛋白质组的人工智能模型有望通过一次简单的血液检测显著提升神经退行性疾病的精准诊断能力。

近年来,神经退行性疾病治疗领域取得了显著进展,包括阿尔茨海默病以及其他疾病的新型治疗手段不断涌现。然而,在临床实践中,疾病的准确诊断仍然面临巨大挑战。
研究人员指出,误诊在痴呆相关疾病中非常普遍,即使在专业医疗机构中误诊率也可达到25%–30%,而在基层医疗环境中甚至可能超过50%。与此同时,多种病理共存是老年群体中的常见现象,尤其是在高龄人群中,多种神经退行性病理同时存在的比例极高。
这种误诊与共病现象不仅影响临床治疗决策,也会干扰新药临床试验的效果评估。因此,开发能够准确识别多种病理的诊断工具成为迫切需求。
血液生物标志物因其易获取、成本低和微创性,成为重要的发展方向。尽管阿尔茨海默病的血液标志物已取得突破,但对于其他神经退行性疾病,仍缺乏可靠的体内诊断指标。
蛋白质组学技术能够一次性检测数千种蛋白,为发现潜在标志物提供了重要机会。然而,这类数据具有高维、非线性和技术噪声等特点,使得传统分析方法难以充分挖掘其价值。
为解决这些问题,研究人员提出结合人工智能与大规模蛋白质组数据,构建能够同时处理多疾病诊断任务的模型,从而提升诊断精度并揭示潜在分子机制。
方法简述
研究人员基于全球神经退行性蛋白质组联盟(GNPC)数据集,构建了一个深度联合学习模型 ProtAIDe-Dx。该模型输入血浆蛋白质组数据,通过多任务学习框架,同时预测六类疾病的诊断结果及其概率。
与传统多分类模型不同,该方法采用联合学习策略,使模型能够同时学习多个疾病任务,从而捕捉不同疾病之间的共享信息,并识别共病情况。
模型输出不仅包括分类结果,还包括低维嵌入表示以及关键蛋白贡献信息,从而支持后续的生物学解释与个体化诊断分析。
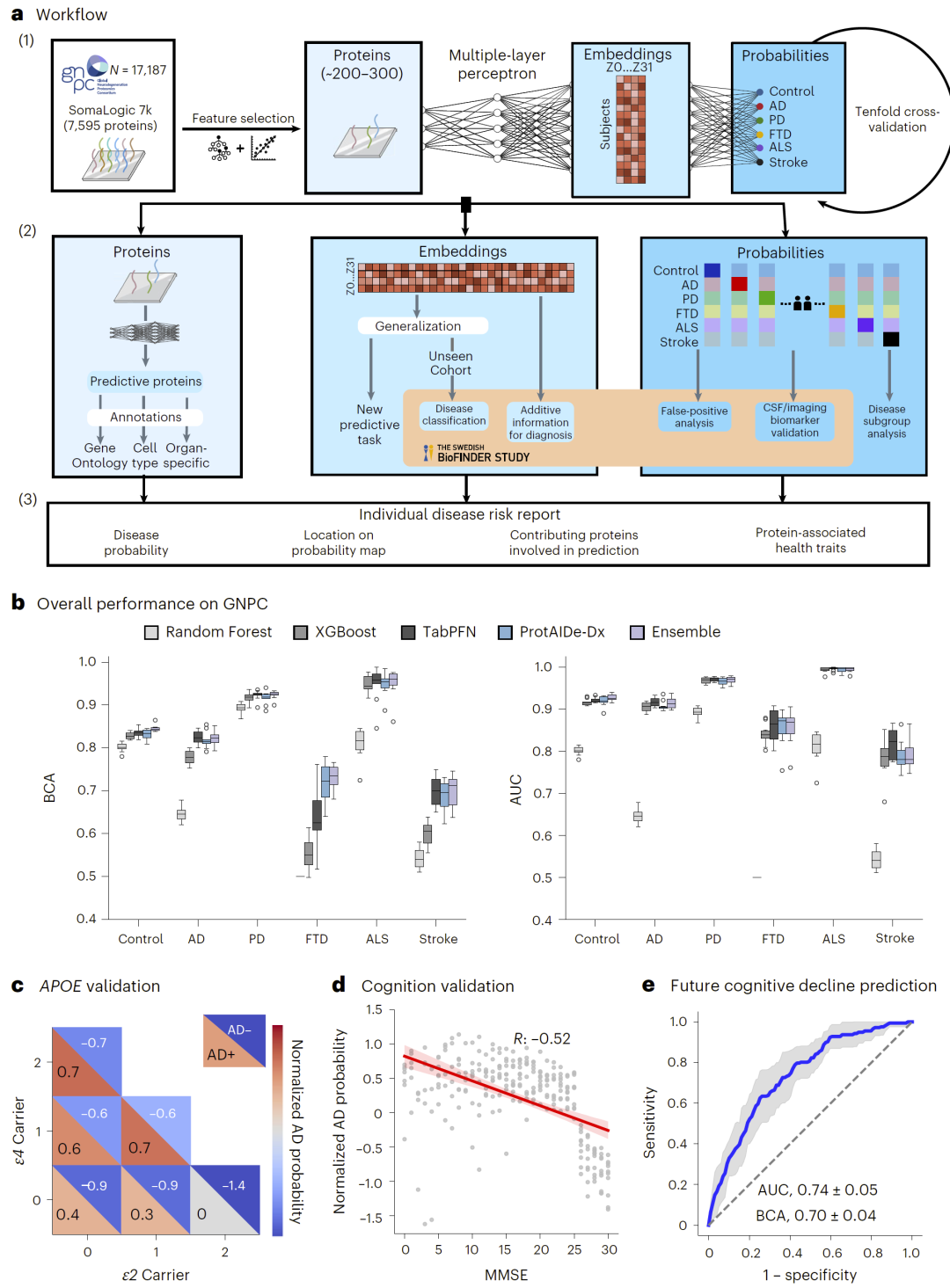
图1:模型整体流程与性能。
结果
联合学习显著提升多疾病诊断能力
研究人员在超过17,000名样本上评估了模型性能。结果显示,ProtAIDe-Dx在所有任务中均表现优于传统机器学习方法。
模型在ALS和帕金森病中的分类准确率超过90%,在阿尔茨海默病和额颞叶痴呆等疾病中也表现出良好的性能。同时,该模型能够在不使用任何临床或人口学信息的情况下,仅依赖蛋白质组数据完成诊断。
进一步分析表明,模型输出的诊断概率与已知风险因素(如APOE基因型)以及认知评分之间具有显著相关性,这表明模型不仅能够进行分类,还能够反映疾病进展程度。
此外,模型学习到的低维蛋白质表示能够推广到新的任务,例如预测未来认知下降风险,显示出良好的泛化能力。
诊断概率揭示疾病异质性与共病特征
通过将个体映射到疾病概率空间中,研究人员发现,不同疾病在该空间中形成清晰分布,但同时也存在交叠区域。
这种交叠反映了不同疾病之间的分子机制重叠,也揭示了潜在的共病现象。例如,一部分阿尔茨海默病患者在空间中更接近脑血管疾病区域,提示其可能存在血管相关病理。
进一步聚类分析发现,每种疾病内部还存在多个亚群,这些亚群在临床特征和蛋白表达模式上存在显著差异。例如,一部分ALS患者表现出更接近FTD的分子特征,并伴随特定基因突变。
这些结果表明,该模型不仅能够进行分类,还能够揭示疾病内部的生物学异质性。
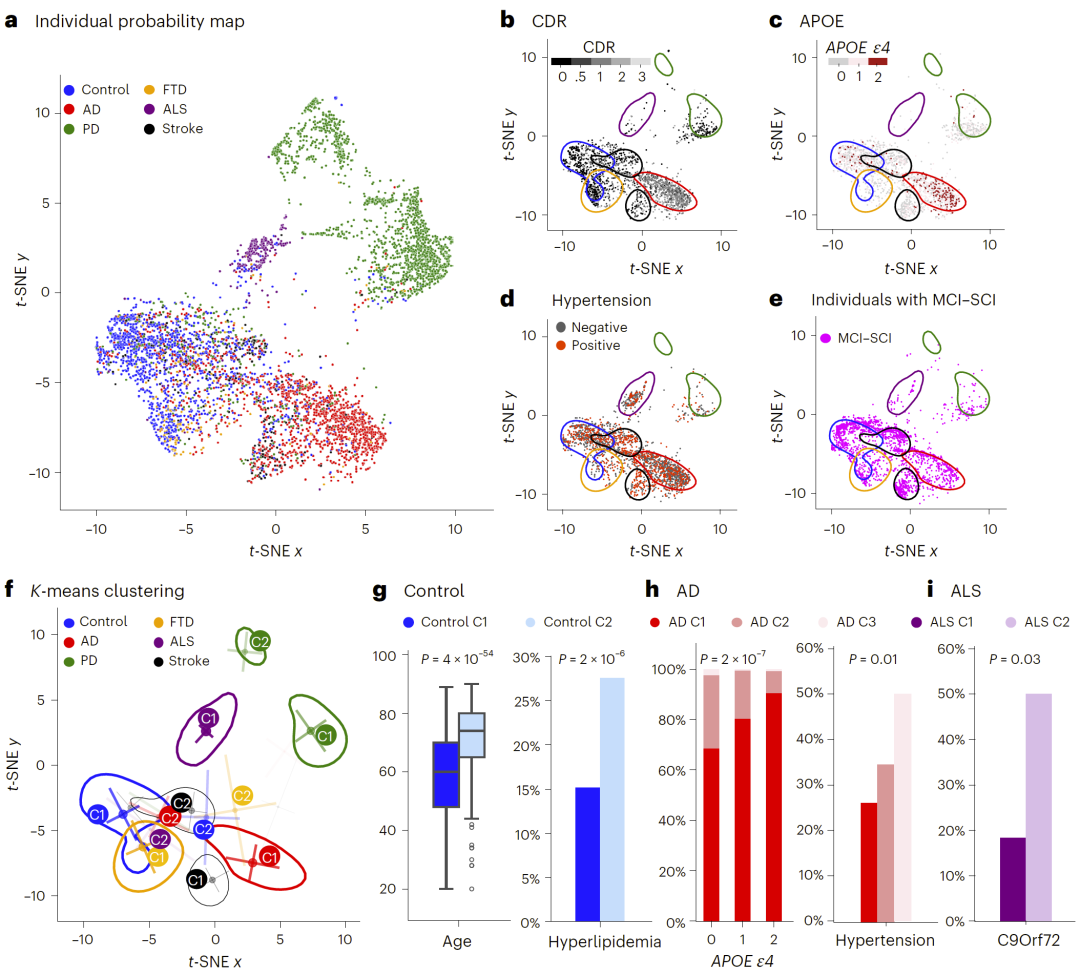
图2:疾病概率空间与异质性。
模型解释揭示关键蛋白与生物网络
了理解模型的决策机制,研究人员分析了对预测最重要的蛋白。
结果显示,模型识别出多个已知与疾病相关的蛋白,同时也发现了新的潜在标志物。例如,不同疾病对应不同的关键蛋白组合,而部分蛋白在多种疾病中均具有重要作用。
进一步分析模型嵌入空间发现,这些低维表示对应不同的生物过程,例如神经功能下降、炎症反应以及血管功能异常等。
这些发现表明,模型不仅具有预测能力,还能够从数据中提取具有生物学意义的分子网络。
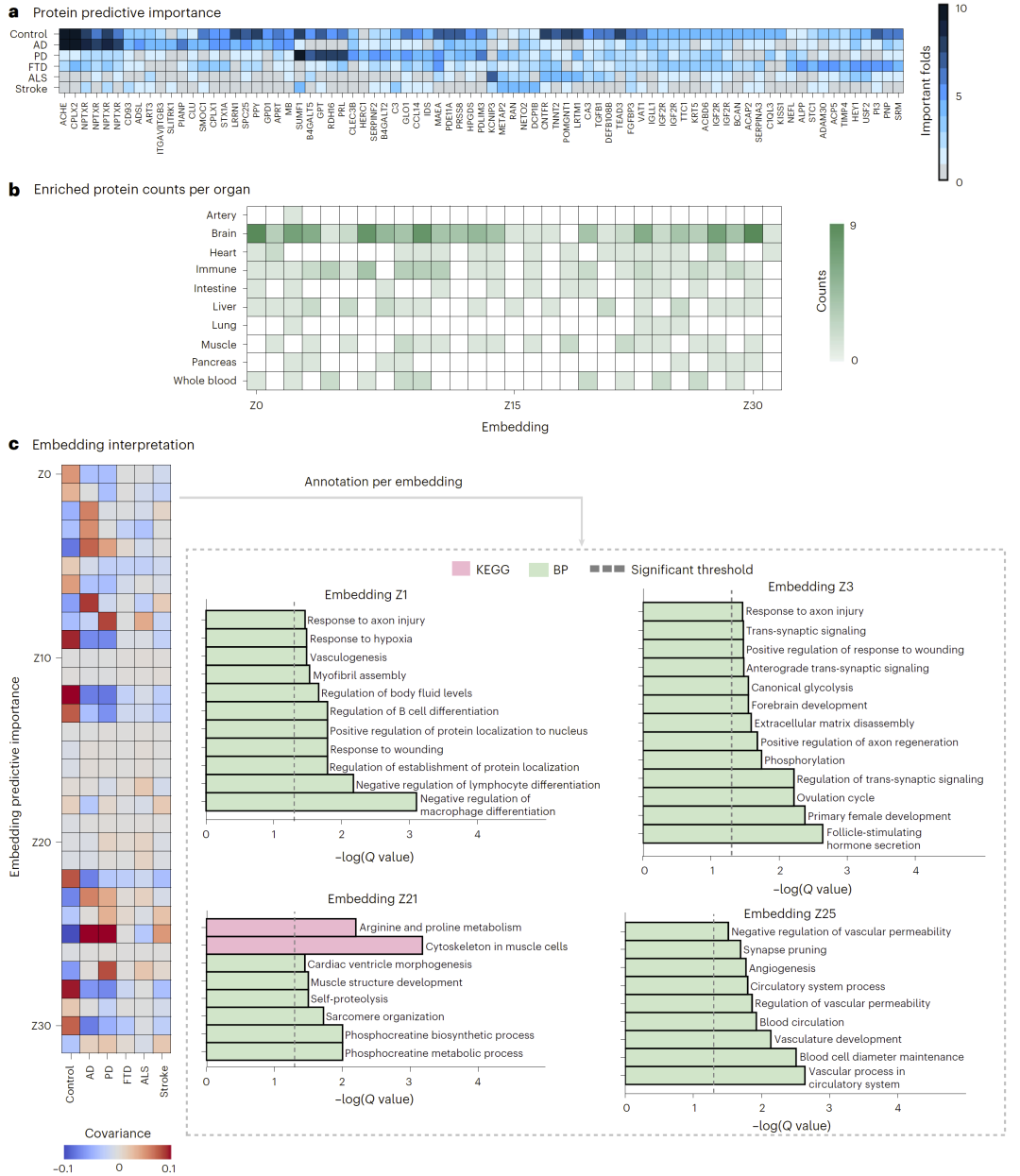
图3:模型解释与蛋白网络。
模型在外部队列中的泛化与验证
研究人员在独立的临床队列中验证了模型性能。尽管不同数据来源之间存在差异,模型仍保持稳定表现。
更重要的是,模型预测概率与多种疾病相关生物标志物显著相关。例如,阿尔茨海默病预测概率与Tau、Aβ等指标一致,脑血管疾病预测概率与白质高信号负担相关。
此外,一些看似“假阳性”的预测,实际上可能反映了尚未临床显现的早期病理变化。
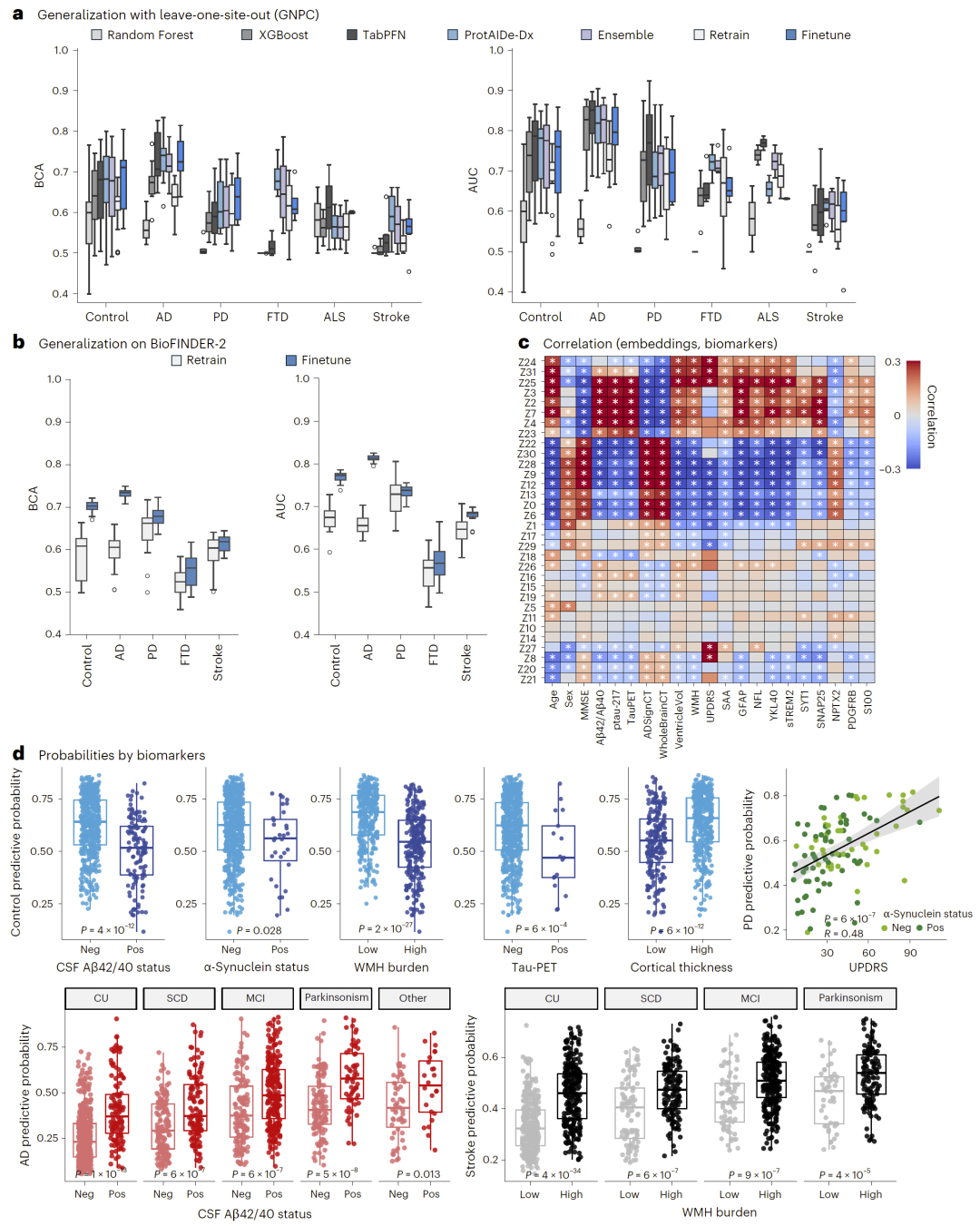
图4:外部验证与生物标志物关联。
蛋白质组信息显著提升临床诊断能力
研究人员进一步评估了该模型在真实临床场景中的应用价值。
结果表明,在结合传统临床指标(如认知评分和影像数据)的基础上,引入蛋白质组模型能够显著提高诊断准确性,尤其是在非阿尔茨海默类痴呆中表现更为明显。
此外,模型预测结果还能有效区分不同患者的认知下降速度,具有潜在的预后评估价值。
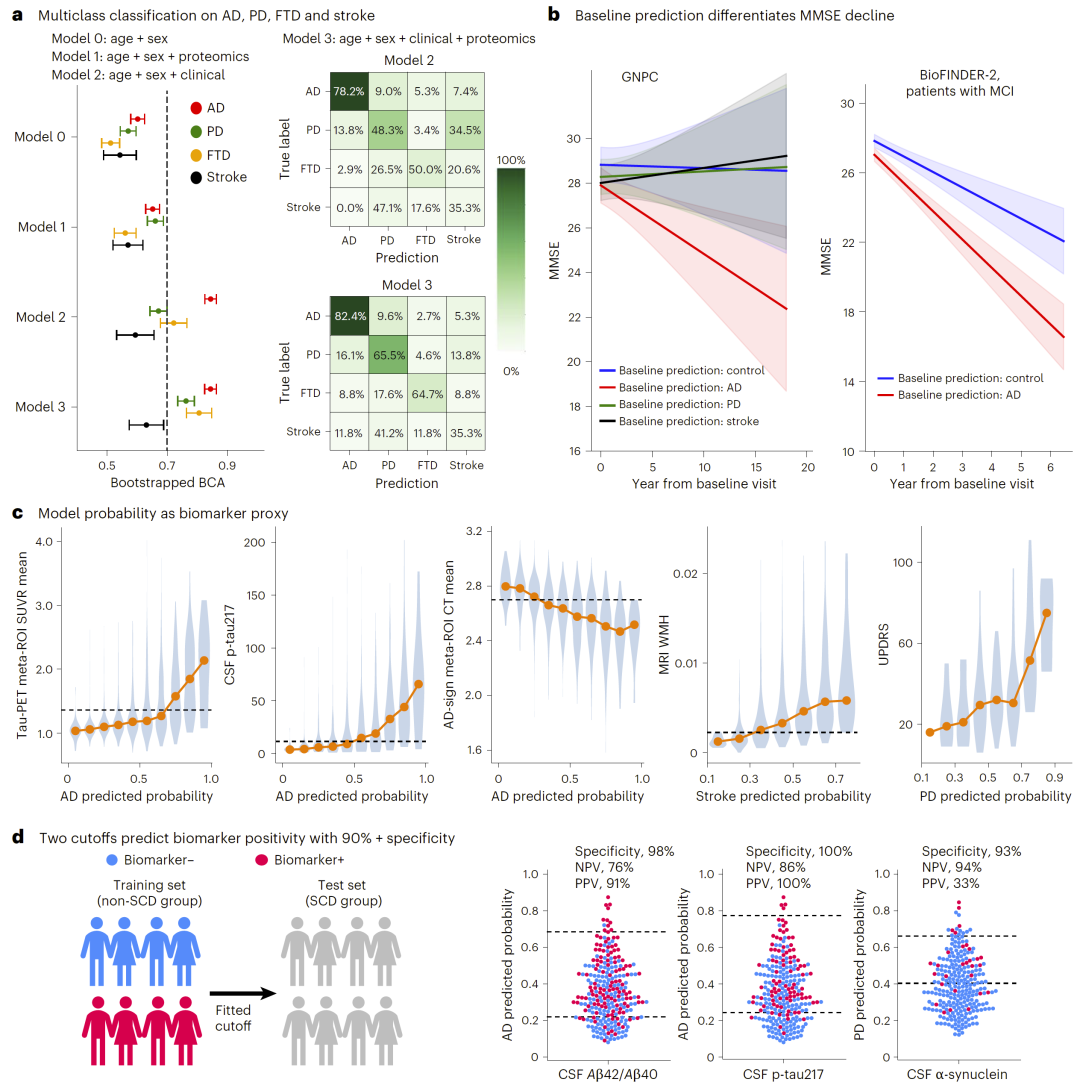
图5:临床应用与增益价值。
模型输出可作为临床生物标志物
研究人员发现,当模型预测概率达到一定阈值时,可高精度预测对应生物标志物状态。
例如,高阿尔茨海默病概率通常对应Tau阳性或皮层萎缩,而高脑血管风险概率则对应更高的白质病变负担。
通过设置双阈值策略,模型在某些任务中可达到超过90%的特异性与预测值,显示出直接应用于临床筛查的潜力。
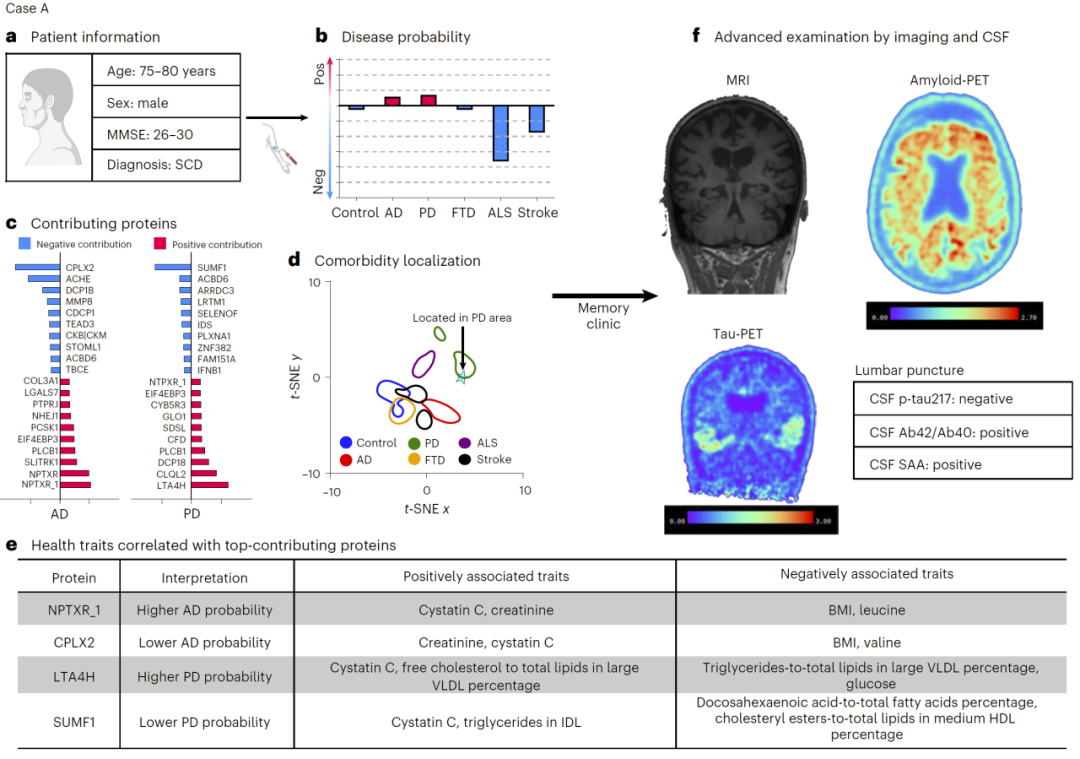
图6:概率阈值与临床决策。
总结
该研究构建了一个基于深度联合学习的蛋白质组模型,实现了对多种神经退行性疾病的同时诊断,并能够识别共病、解释分子机制以及提升临床决策能力。
这一工作的重要意义在于:
通过一次血液检测,即可获得多疾病、多层次的信息,从而推动神经退行性疾病诊断从单病种、单标志物向系统化、个体化方向发展。
整理 | DrugOne团队
参考资料
An, L., Pichet Binette, A., Hristovska, I. et al. A deep joint-learning proteomics model for diagnosis of six conditions associated with dementia. Nat Med (2026). https://doi.org/10.1038/s41591-026-04303-y

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢