

一、概要
同源性检索是现代分子生物学识别序列祖先与功能的基石,经历了从BLAST等传统启发式搜索到Foldseek等结构比对方法的演进。然而,该领域仍面临两大核心挑战:一是生物序列数据量极其庞大,其中宏基因组数据库规模高达十亿级;二是高精度检索(尤其是蛋白结构比对和长核苷酸检索)计算成本高昂且速度缓慢。针对这些瓶颈,研究者提出了基于生物语言大模型的检索增强型比对搜索工具——ERAST,相关工作发表在Nature旗舰期刊《自然·生物技术》(5-year IF: 59.5)。
ERAST通过集成ESM2预训练蛋白质语言模型(Protein-LLM)与Caduceus长序列预训练核苷酸语言模型(Nucleotide-LLM),将海量生物序列编码为高维向量,从而构建了当前规模最大、涵盖超过10亿条的生物序列向量数据库。ERAST利用预训练生物语言大模型所习得的进化、结构与功能特征,增强序列向量表示质量,提升检索后重排序效率,拓展分布外序列的检索泛化能力,为大规模生物分子同源性搜索提供了全新的高效解决方案。
该文章共同第一作者为浙江大学计算机学院研究生姜一诺和腾讯AI Lab研究员何冰,通讯作者为浙江大学计算机学院求是特聘教授陈华钧、浙江大学国际联合学院百人计划研究员张强及腾讯AI Lab医疗首席科学家姚建华。
二、方法
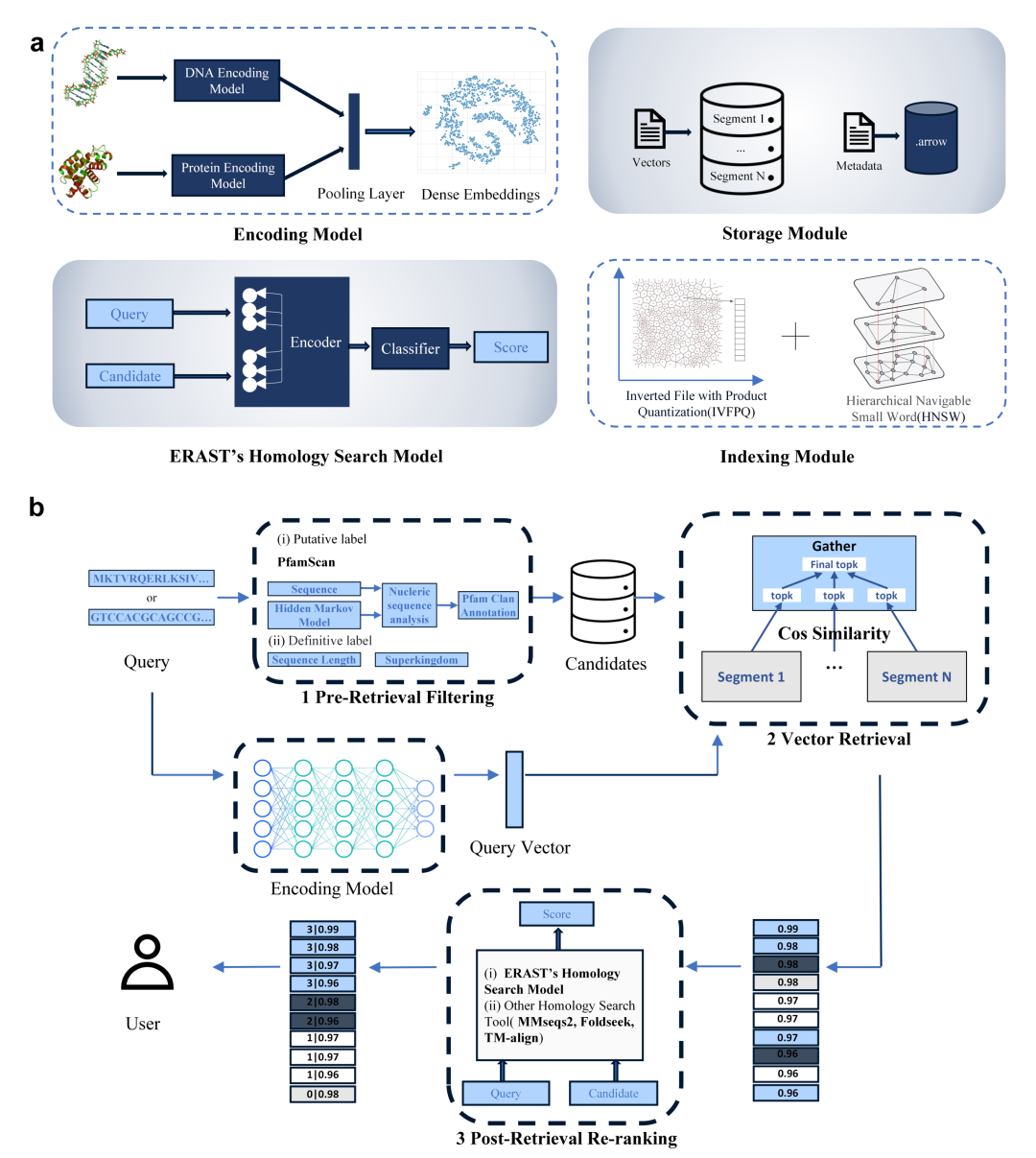
2.1 检索流程
(1)检索前过滤: 此阶段的核心是通过添加元数据(即描述向量嵌入内容的额外字段)来优化原始查询,从而过滤无关数据。基于具体元数据的过滤可以缩小搜索空间,显著提升同源性检索质量。例如,蛋白质的元数据可包括序列长度或 Pfam 家族标签;核苷酸序列则可包括超界(superkingdom)等分类标签。这种机制让 ERAST 能更高效地锁定相关候选目标。
(2)向量检索: 接收查询后,ERAST 使用与建库时相同的模型将其转化为向量,并计算其与过滤后候选向量的相似度(基于余弦距离)。为进一步加速大规模搜索,系统采用了多线程策略,对数据库分段进行并行检索和候选筛选,最后汇总各段结果并按相似度得分择优。
(3)检索后重排序: 针对远源同源序列的精准检索仍是挑战。以往研究侧重于改进编码模型,但每次模型更新都要重新编码千万级序列,成本极高。ERAST 引入了检索后重排序机制,通过设计的评分模型 (EHSM) 评估查询序列与目标的匹配度并重新排序。与侧重表征的编码模型不同,该评分模型的目标是学习如何进行排序。我们将查询序列与每个候选序列拼接后输入 EHSM,并将同源性评分建模为一个多标签分类任务。具体而言,模型根据候选蛋白质与查询蛋白质的关系亲疏进行评分,将其分为四个等级:(1) 同族;(2) 非同族但同超家族;(3) 非同超家族但同折叠类型;(4) 非同折叠类型。该重排序模块具有可插拔性,支持灵活集成 EHSM、MMseqs2 或 TM-Align 等多种策略,在不频繁更新向量库的前提下提升检索精度。
2.2 基于预训练语言模型的向量数据库构建
首先,利用预训练语言模型将原始序列转换为连续的向量表征。在蛋白质方面,经对比多个模型后,选择 ESM2 作为编码模型以增强对远源同源蛋白质的召回率,并采用平均池化层提取最终特征。针对长度超过 10,000 bp 的核苷酸序列,采用基于MAMBA 架构的 Caduceus 长文本核苷酸预训练DNA语言模型作为骨干架构,并利用遗传分类数据对预训练获得的 DNA 模型进行后训练,作为核苷酸序列的编码器。
为在亿级稠密向量库中实现快速精准检索,系统采用了平衡精度与速度的索引算法。具体而言,选择了 IVFPQ(倒排文件乘积量化)结合 HNSW(分层导航小世界)算法来组织和存储向量。为解决扩展性问题并缩短响应时间,数据库根据 UniProt 原始数据的长度顺序进行了分段(每段 1000 万个样本),并为每段独立构建索引。此外,元数据采用 Arrow 格式存储,以支持大规模异构数据集的高效流式传输。最终,我们构建了一个涵盖超过 10 亿条生物序列的向量数据库,数据来源包括:UniParc (8.07 亿蛋白质序列),UniRef90 (1.90 亿蛋白质序列),UniRef50 (5910 万蛋白质序列),Swiss-Prot (57.2 万蛋白质序列),RefSeq (3480 万核苷酸序列)。
2.3 蛋白功能聚类
UniRef50 和 UniRef90 是通过利用 MMseqs2 基于局部相似性对 UniRef100 序列进行聚类构建的,而我们则是通过 ERAST 对 UniRef50 和 UniRef90 应用全局相似性聚类来构建功能簇,这展示了 ERAST 有助于阐明蛋白质功能的潜力。我们通过 ERAST 将网络范围扩展至整个 UniRef90 数据库,最终得到 8,841,389 个功能簇。在 UniRef90 中所有未知功能的暗簇中,94% 在生成的网络中与已知功能的亮 UniRef90 簇相连,揭示了超过 1.58 亿种独特蛋白质的潜在进化关系。
三、实验分析
3.1 同源性检索评估
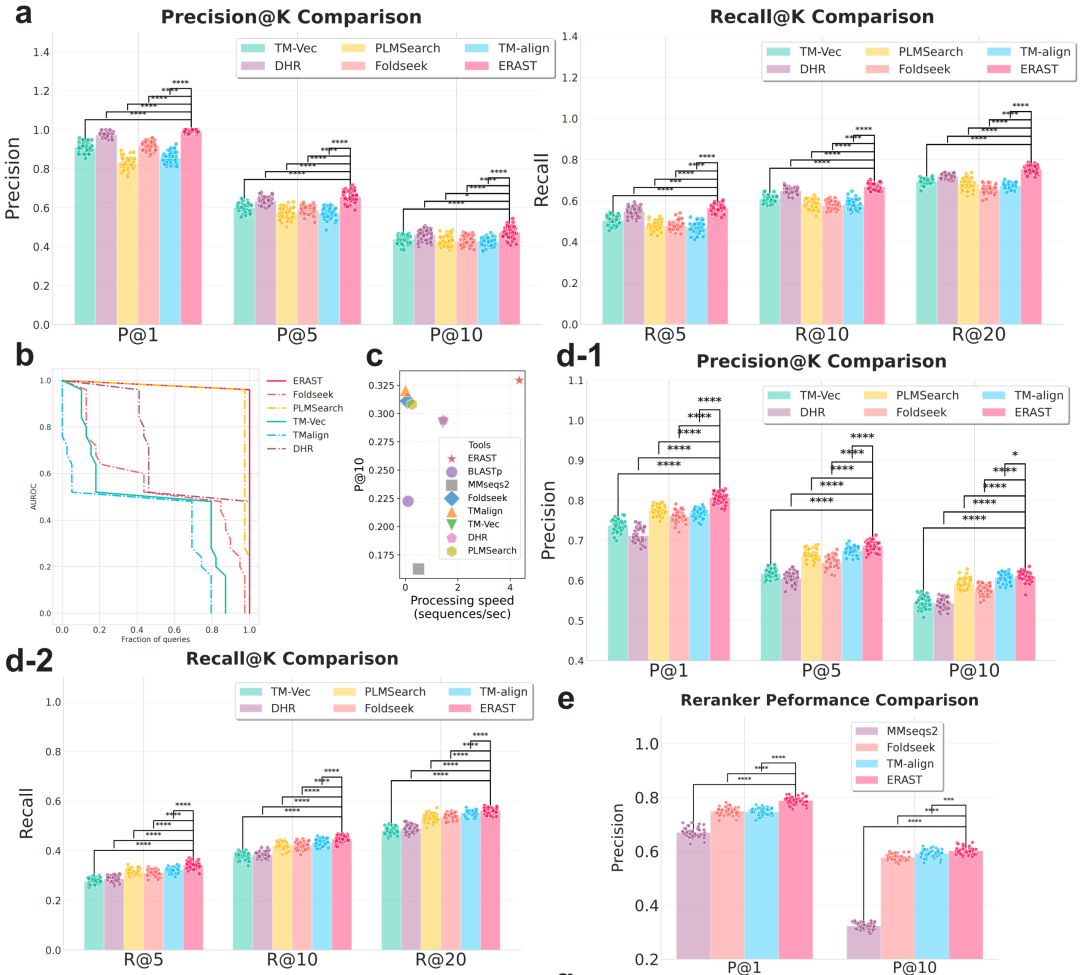
我们对比了序列搜索(MMseqs2、Blastp)、结构字母法(Foldseek等)、结构比对(TM-align等)及深度学习方法(DHR等)。在 SCOPe40 测试集上,ERAST 的 P@1 分别比 TM-Vec 和 PLMSearch 高出 6.9% 和 2.9%。在检索速度上,不含重排序阶段的 ERAST 比主流工具 Foldseek 快 50 倍。在分布外序列测试中,ERAST 表现出极强的鲁棒性,AUROC 比在多领域注释上表现不佳的 TM-align 高出 62.2%。进一步的消融表明,预训练蛋白质语言模型虽能捕捉家族级结构,但在区分折叠级(Fold level)时存在困难。ERAST 通过检索前过滤提升了整体结果质量,并通过检索后重排序(EHSM)进一步优化了顶部结果,使平均 P@1 从 0.741 提升至 0.821。针对核苷酸序列,我们将检索视为分类任务,对比了 Blastn、Kraken2 等工具。相较于其他方法,ERAST 展现出一致的优势,其属级(Genus level)AUROC 提升至 0.186,远超第二名。与最常用的 MMseqs2 相比,ERAST 在界、门、属等级别的平均精度分别提升了 32.4%、18.1% 和 14.3%。
3.2 检索速度评估
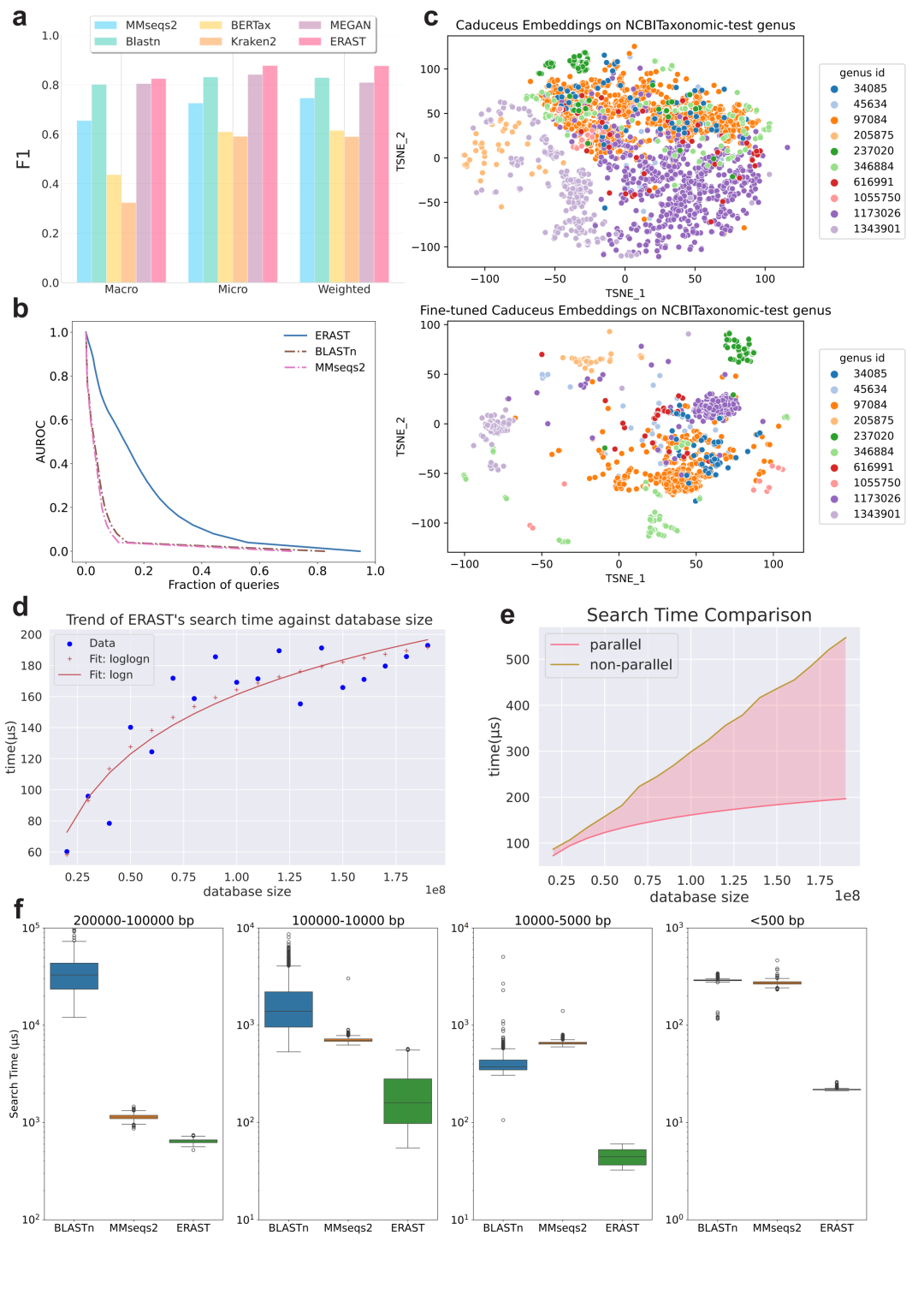
借助稠密向量与索引量化技术,ERAST 在由 UniParc、UniRef90、UniRef50 及 RefSeq 构成的十亿级向量数据库上实现了毫秒级检索。在蛋白质检索实验中,ERAST 在相同计算资源下(64核 CPU)对比发现,其无重排序版本在速度与精度上均优于现有的深度学习及结构比对方法:其检索速度比 Foldseek 快 50 倍,比 TM-align 快 5 万倍,且 P@10 精度更高。针对长核苷酸序列,通过将序列映射为固定维度的稠密向量,ERAST 在各长度区间(从 500bp 以下到 10 万 bp 以上)均保持最快速度;当序列超过 10 万 bp 时,其速度约是 BLASTn 的 60 倍、MMseqs2 的 2 倍。通过在 Swiss-Prot 和 UniRef90 子集上的扩展性测试证实,采用并行搜索策略使 ERAST 的时间复杂度从 O(n) 降至 O(log n) 到 O(loglog n) 之间,从而实现在毫秒内完成对超过 10 亿规模数据库的查询。
四、结论
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢