
DRUGONE
药物分子的结构优化,尤其是提升其针对特定靶点的活性,是化学与药物研发中的核心难题之一。研究人员利用化学语言模型在序列数据学习方面的优势,提出了一种模拟真实药物研发过程的训练策略。该方法通过逐步引入活性逐渐增强的分子,对模型进行增量式微调,使其在生成分子时自然偏向高活性结构。
研究表明,这种渐进式训练策略可以有效引导模型生成具有更高生物活性的类似物,甚至在没有外部评分函数的情况下,超越已知分子的活性水平。进一步分析发现,模型不仅能够捕捉结构–活性关系(SAR),还能够理解分子中的长程依赖关系,从而实现基于已有知识的结构优化设计。这些结果表明,化学语言模型在药物分子结构优化任务中具有重要潜力。

化学语言模型通过处理SMILES字符串,实现对分子结构的学习与生成。在大规模分子数据上预训练后,这类模型能够掌握化学语言的语法规则及基本性质,并通过迁移学习,从少量数据中提取特定任务相关信息,例如生物活性。
这类模型已在从头药物设计中取得成功,能够生成具有目标活性的分子。然而,相比“生成新分子”,更具挑战性的任务是结构优化——即在已有分子骨架基础上,提高其对特定靶点的活性。
传统实验方法依赖反复的“设计–合成–测试”循环,成本高且耗时长。计算方法虽提供一定帮助,但现有生成模型在该任务上仍存在明显局限。例如,多数方法依赖外部评分模型(如QSAR或分子对接),而这些“外部评价器”本身的不确定性会影响整体性能。
此外,很多研究只优化简单性质(如logP或QED),无法反映真实药物设计的复杂性。因此,如何在仅依赖配体信息、无需外部评分的情况下实现活性优化,仍是未解决的问题。
研究人员提出,通过模拟药物研发中的知识积累过程,将活性信息作为“时间序列”引入模型训练,从而解决这一难题。
方法
研究人员构建了一种渐进式微调策略。模型首先在大规模分子数据上预训练,并刻意去除目标相关分子,使模型对目标“无先验认知”。随后,将具有结构–活性关系的数据按活性排序,分阶段输入模型进行训练。
在每一阶段,模型接触到的是活性更高的一组分子,这一过程类似于药物研发中逐步优化的路径。通过这种方式,模型不仅学习分子结构,还学习“如何变得更优”。
在评估方面,研究人员设计了一种“再发现评分”,用于衡量模型是否能够生成未见过但高活性的分子。同时,通过perplexity分析模型对分子的偏好变化。该方法在两个靶点上进行了验证:PPARγ和RORγ。
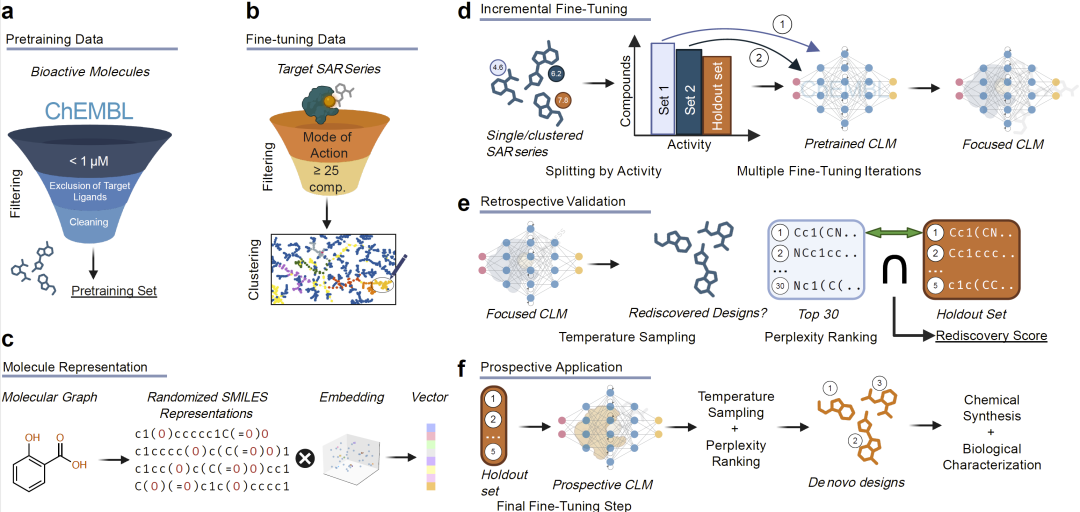
图1: 化学语言模型(CLMs)及其在本研究中用于分子结构优化的应用框架。
结果
渐进式训练显著提升模型设计高活性分子的能力
研究人员首先在PPARγ体系中进行回顾性验证。结果显示,相比传统一次性训练,渐进式微调显著提高了模型“再发现”高活性分子的能力。
模型在逐步学习过程中表现出更高的相似性和更强的目标聚焦能力。同时,最佳训练策略为将数据分为4–5个阶段逐步输入。
值得注意的是,这种提升与数据规模、分子多样性无关,说明方法具有较强的普适性。
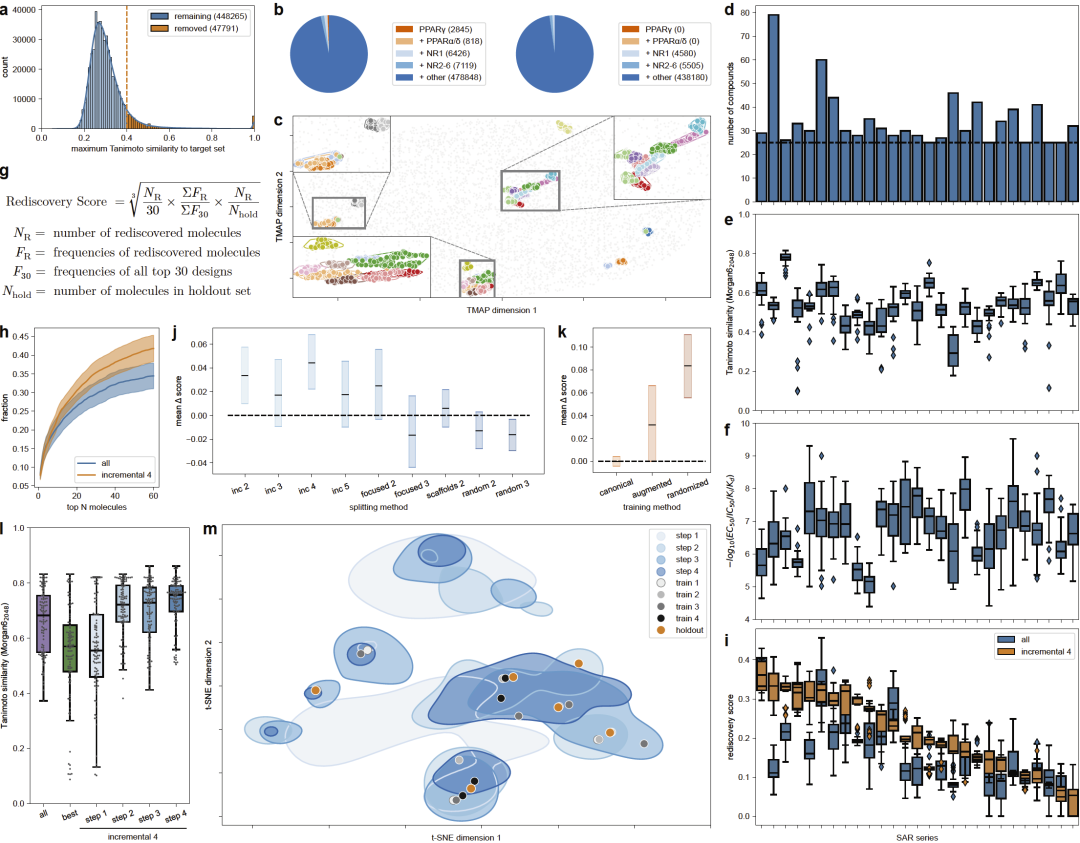
图2: 渐进式CLM训练的回顾性评估。
模型成功设计出超越已知分子的PPARγ激动剂
在前瞻性实验中,研究人员选择一类PPARγ激动剂进行测试。模型生成的分子中,绝大多数保持了目标骨架结构,并在关键取代基上进行了优化。
合成并测试的9个分子全部表现出高活性,其中多个分子的活性超过已知最优分子,部分提升达到数十倍。
这些结果表明,模型不仅复制已有知识,还能“外推”并设计更优结构。
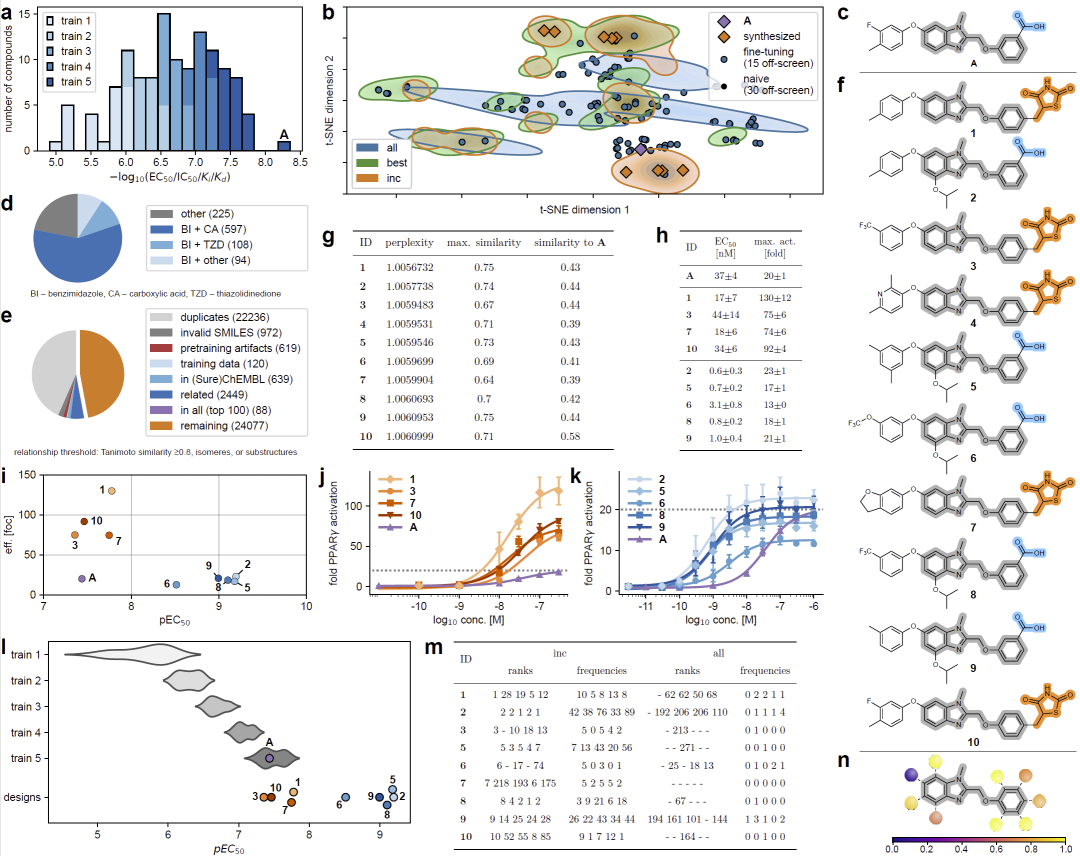
图3:基于渐进式CLM训练的PPARγ激动剂结构优化。
方法可泛化至RORγ体系并揭示复杂SAR规律
在RORγ体系中,研究人员同样观察到模型能够生成高活性分子。其中一个设计分子活性显著优于训练集中最相似分子。
更重要的是,模型识别出了关键结构片段(如tert-butyl基团)对活性的影响,并能理解其“上下文依赖性”,即该基团仅在特定结构背景下才有效。
这表明模型具备捕捉复杂SAR关系的能力。
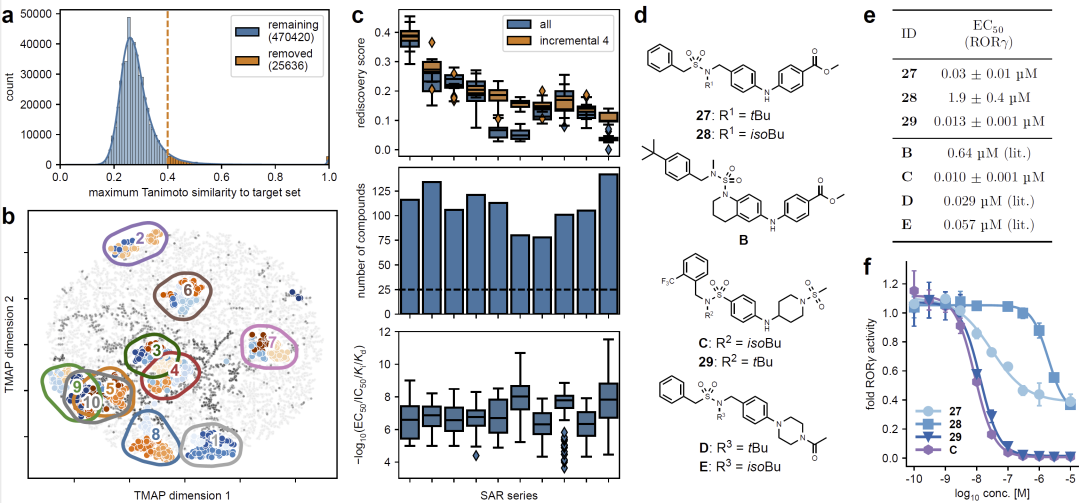
图4: 基于渐进式微调CLM的RORγ反向激动剂设计。
渐进式训练改善模型对SAR的理解
进一步分析表明,渐进式训练显著改变了模型内部表示。相比传统训练方式,该方法使模型的perplexity与分子活性呈正相关,即模型更倾向于生成高活性分子。
同时,模型学习过程呈现出“阶梯式提升”,反映出逐步引入高质量数据的优势。
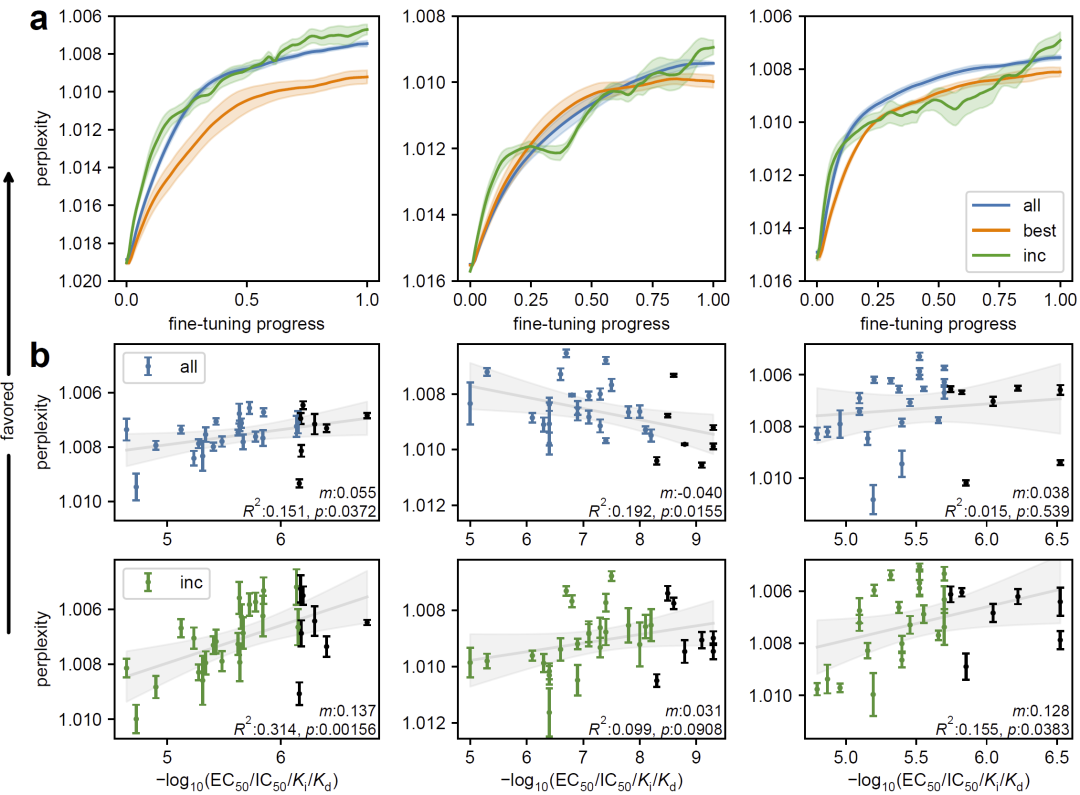
图5: 渐进式微调提升模型在高活性分子上的困惑度表现及整体活性–困惑度相关性。
讨论
研究表明,药物分子结构优化可以通过纯数据驱动方式实现,而无需依赖外部评分函数或蛋白结构信息。这一突破的关键在于引入“学习轨迹”,使模型能够像药物化学家一样逐步积累经验。
渐进式训练的优势可能来源于更平滑的优化路径,以及递归神经网络对序列信息的天然适应能力。通过将活性排序作为训练维度,模型能够更高效地提取关键结构特征。
在PPARγ和RORγ两个体系中的成功应用表明,该方法既适用于局部结构优化,也适用于更广泛的SAR迁移任务。
总体而言,这项工作拓展了化学语言模型在药物设计中的应用边界,使其不仅能“生成分子”,还能够“优化分子”,在真实药物研发流程中具有重要潜力。
整理 | DrugOne团队
参考资料
Hörmann, T., Mayer, D., Lewandowski, M. et al. Structural optimization of drug molecules with incrementally trained language models. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-71591-w

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢