
近日,华东师范大学计算机科学与技术学院张倩副研究员团队与华东理工大学药学院徐志建教授团队合作,在生物信息学领域国际期刊 Briefings in Bioinformatics 发表题为 “PEPNet: a two-stage point cloud framework with hierarchical embedding and antigen–antibody interaction modeling for epitope prediction” 的研究论文。该研究提出了一种融合层级嵌入与抗原–抗体相互作用建模的两阶段原子级3D点云表位预测网络 PEPNet,为构象表位精准识别、治疗性抗体开发和疫苗设计提供了新的计算工具。

背景介绍
B细胞表位是抗体识别抗原并触发免疫反应的关键区域,其准确预测对亚单位疫苗设计、治疗性抗体开发和疾病诊断具有重要意义。然而,表位预测仍面临重要挑战。多数B细胞表位属于构象表位,即相关氨基酸残基在一级序列上可能并不连续,但在蛋白质三维折叠后形成空间邻近区域。这使得仅依赖序列信息或残基层级结构表示的方法难以充分捕捉抗原–抗体识别中的精细空间特征。
为什么需要抗体感知的表位预测?
需要说明的是,构象表位预测可以分为两类任务。一类是抗体无关的潜在表位预测,即在尚无特定抗体信息时,仅根据抗原结构判断哪些区域可能成为抗体识别位点。这类方法主要用于疫苗抗原设计、免疫原区域筛选和潜在中和表位发现。另一类是抗体感知的配对特异性表位预测,即在已有抗体结构、序列或抗原–抗体复合物结构信息时,判断某一特定抗体究竟识别抗原上的哪些残基。这类任务更适用于治疗性抗体开发、抗体优化、逃逸突变分析和作用机制解析。PEPNet 聚焦后一类问题。
现有方法的核心瓶颈
现有结构建模方法多采用残基层级图表示,通常通过距离阈值构建残基间连接。残基层级图表示会将连续的三维空间离散化,可能导致抗原–抗体识别所需的高分辨率几何特征、方向信息和局部相互作用细节流失。蛋白质结构文件本身天然包含原子级三维坐标。与残基层级图表示相比,原子级三维点云可以更直接地保留蛋白质真实的空间几何形状。因此,研究团队提出原子点云表位预测网络PEPNet,将蛋白质建模为原子级三维点云,用于更精细地刻画抗原–抗体识别过程。不过,直接将计算机视觉领域的三维点云方法迁移到蛋白质体系并不简单,主要面临三方面挑战:一是常见的点云局部聚合策略,如 FPS 和 KNN,往往按照几何空间进行任务无关划分,忽视了蛋白质中“原子组成残基”的天然生物层级;二是点云在数学上是无序集合,而蛋白质序列具有严格的化学方向性和生物学顺序;三是现有抗原–抗体复合物数据集规模有限,难以支撑复杂深度学习模型的充分训练。
PEPNet 的核心创新
针对上述挑战,PEPNet 从结构表示、生物学先验、序列顺序建模、数据高效利用和抗原–抗体相互作用建模五个方面进行了系统设计(图1)。
首先,PEPNet 将蛋白质表示为原子级三维点云。每个原子被视为空间中的一个点,并携带原子类型、局部表面法向量等理化属性。相比传统残基图表示,这种方式能够更直接地保留蛋白质结构中的高分辨率空间几何信息和物理连续性。
其次,PEPNet 设计了残基感知层级嵌入模块(Residue-aware Hierarchical Embedding Module, RHEM,图2)。该模块显式利用蛋白质中“原子—残基”的天然层级关系,将原子级特征聚合为残基级表示。与简单拼接原子特征和残基特征不同,RHEM 能够在局部构建更具生物学意义的结构单元,使模型在保留精细几何信息的同时,形成符合蛋白质结构规律的残基表示。
第三,PEPNet 引入旋转位置编码(Rotary Positional Encoding, RoPE),将蛋白质序列顺序信息注入点云表示。点云方法通常具有排列不变性,即打乱输入点的顺序不影响输出结果;但对于蛋白质而言,残基顺序决定肽链连接方式、空间折叠和生物学功能。RoPE 通过对注意力机制中的 Query 和 Key 向量进行旋转变换,使模型能够在三维几何表示中感知残基序列的相对顺序。
第四,PEPNet 采用BERT 风格的自监督预训练策略和多样化数据增强,以缓解抗原–抗体标注数据不足的问题。在预训练阶段,模型随机遮蔽部分抗原或抗体残基,并通过多任务学习重建氨基酸类型、溶剂可及性和 PSSM 矩阵等理化属性,从而在有限数据条件下学习蛋白质内在结构规律。与此同时,研究团队还引入高斯噪声、随机三维旋转等数据增强策略,提高模型对结构噪声和预测结构的鲁棒性。
在微调阶段,PEPNet 进一步引入抗体感知的交叉注意力解码器。该模块能够实现从抗体到抗原的残基级信息流动,显式建模抗原–抗体之间的结合相互作用,从而预测特定抗体识别的抗原残基。
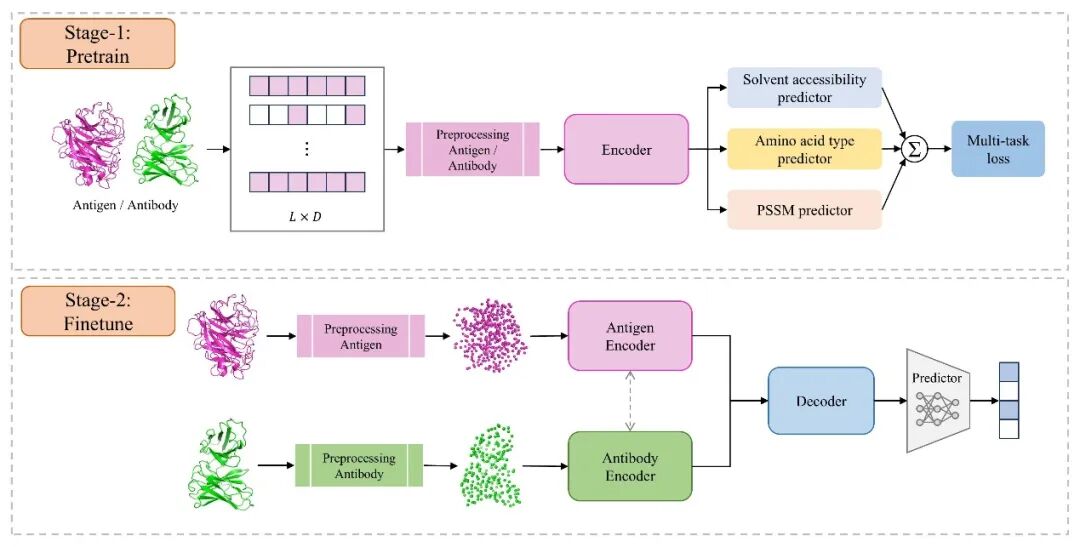
图 1: PEPNet流程示意。
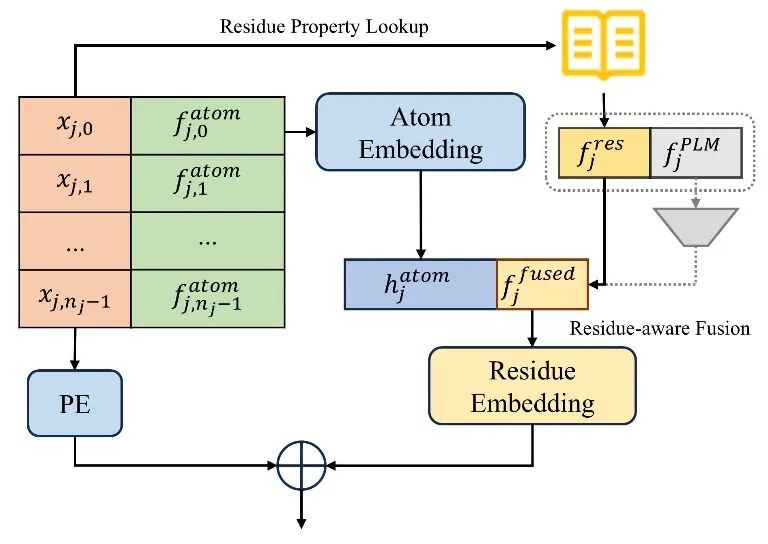
图 2: PEPNet 中的残基感知层级嵌入模块。
性能表现
研究团队在 AsEP 数据集上对 PEPNet 进行了系统评估,并与多种代表性方法进行比较,包括序列方法、结构方法和多模态方法(表1)。
在按照表位与抗原表面残基比例划分的 Ratio 设置下,PEPNet 在 MCC、AUC、Precision 和 F1 等关键指标上表现突出,MCC 达到 0.401,AUC 达到 0.765,整体优于所有对比方法。相较于代表性多模态方法 WALLE,PEPNet 在 MCC 和 AUC 等指标上均取得明显提升。
值得注意的是,当使用 AlphaFold3 预测结构进行评估时,PEPNet 仍保持较好的鲁棒性,MCC 达到 0.346,与WALLE相比,PEPNet将MCC提高了13.4%,AUC提高了8.1%。这说明 PEPNet 不仅适用于实验解析结构,也具备应用于预测结构的潜力,为真实场景下的抗原–抗体配对分析提供了可行路径。
在更具挑战性的 Group 设置下,PEPNet 结合蛋白语言模型嵌入后(PEPNet+LE),在 MCC、AUC 和 Precision 等指标上同样取得领先表现,说明该框架具有较好的泛化能力。
表 1 对比实验结果

案例分析
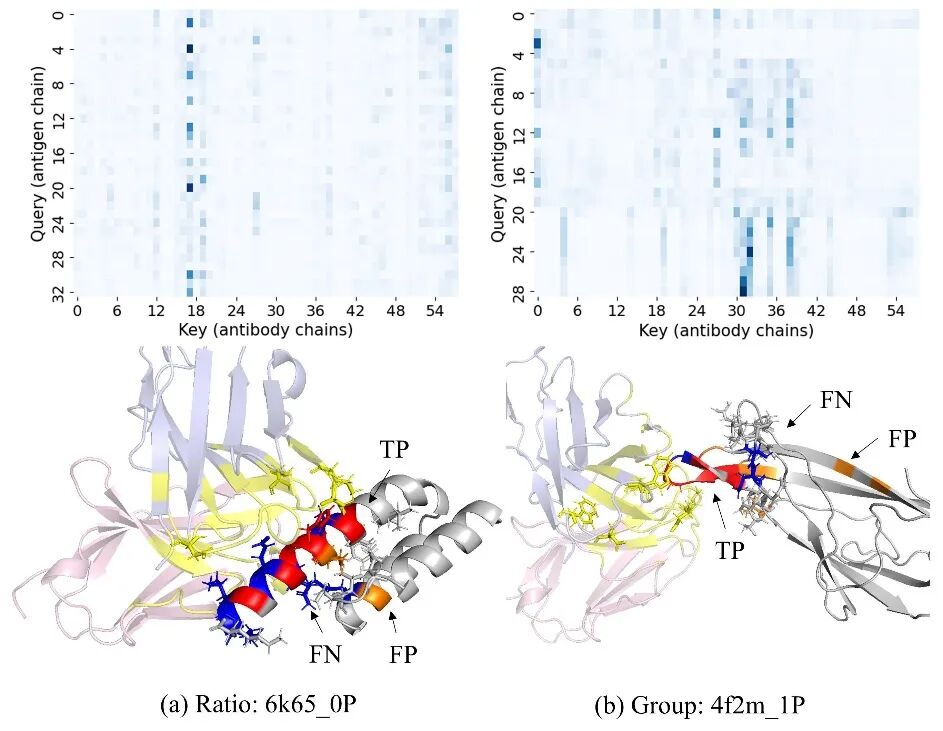
图 3 PEPNet与PEPNet+LE对代表性抗原-抗体对进行表位预测的结果。
在案例分析中,研究团队选取了两个代表性抗原–抗体复合物进行可视化(图3)。结果显示,模型预测过程中赋予高注意力分数的抗原残基与实验注释表位高度重合;同时,高注意力分数的抗体残基主要集中于抗体环区,这与抗体通过互补决定区识别抗原的生物学机制相一致。这表明 PEPNet 不仅能够提升预测性能,也能够为抗原–抗体相互作用机制分析提供一定的可解释性线索。
该论文第一作者为华东师范大学硕士研究生陈佳怡,通讯作者为华东理工大学药学院特聘教授徐志建和华东师范大学计算机科学与技术学院副研究员张倩。该工作还得到了华东师范大学计算机科学与技术学院院长张桂戌教授的大力支持。
参考资料
Jiayi Chen, Guixu Zhang, Zhijian Xu, Qian Zhang, PEPNet: a two-stage point cloud framework with hierarchical embedding and antigen–antibody interaction modeling for epitope prediction, Briefings in Bioinformatics, Volume 27, Issue 1, January 2026, bbag067
https://doi.org/10.1093/bib/bbag067
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢