

生物语言模型(Biological language model)的性能高度依赖于预训练数据的质量、多样性和规模。尽管宏基因组数据集(metagenomic datasets)具有巨大的生物多样性,但由于数据可访问性、质量过滤和去重方面的挑战,其作为预训练数据的利用一直受到限制。本文介绍了开放宏基因组(Open MetaGenomic,OMG)语料库,这是一个包含3.1T个碱基对和3.3B个蛋白质编码序列的基因组预训练数据集,它整合了两个最大的宏基因组数据集存储库(JGI的IMG和EMBL的MGnify)。我们首先记录了数据集的组成,并描述了为去除低质量数据而采取的质量过滤步骤。我们将OMG语料库作为混合模态基因组序列数据集提供,该数据集表示多基因编码的基因组序列,其中包含蛋白质编码序列的翻译氨基酸和基因间序列的核酸。我们训练了第一个混合模态基因组语言模型(gLM2),该模型利用基因组上下文信息来学习蛋白质-蛋白质相互作用界面中稳健的功能表征和共进化信号。此外,我们证明,嵌入空间中的去重可以用于平衡语料库,从而提高下游任务的性能。

论文:The OMG dataset: An Open MetaGenomic corpus for mixed-modality genomic language modeling
单位:美国Tatta Bio公司、美国劳伦斯伯克利国家实验室、欧洲分子生物学实验室、韩国首尔国立大学生物科学学院、美国麻省理工学院
发布日期:2025年
OMG dataset https://huggingface.co/datasets/tattabio/OMG
gLM2 https://huggingface.co/tattabio/gLM2_650M
请索引第85篇论文
 |  |
打破AI生物学壁垒!ICLR 2025开源3.1T海量数据集,混合模态gLM2实现降维打击
当AI遇上生物学,我们总在感叹“巧妇难为无米之炊”。高质量的生物数据到底在哪里?最近ICLR 2025收录的一篇重磅论文给出了答案。来自麻省理工学院(MIT)、伯克利国家实验室等顶尖机构的研究团队,开源了一个高达3.1T碱基对的宏基因组预训练数据集(OMG),并基于此训练了能同时处理核酸与蛋白的混合模态模型gLM2。这不仅是一场数据规模的狂欢,更是一次模型架构与预训练思路的降维打击!
直击痛点:为什么我们需要宏基因组(Metagenomic)?
在“AI for Science”浪潮中,生物语言模型(Biological Language Models)的性能往往被预训练数据的质量与多样性所限制。过去,大家习惯使用像UniProt这样的“精品题库”来训练模型(如著名的ESM系列)。但自然界中存在着海量的“未知微生物”,这些未被培养、未被标注的序列才是真正的宝藏。
遗憾的是,宏基因组数据虽然庞大,却因为下载困难、组装质量参差不齐、难以去重等问题,一直未能成为AI模型的主流“口粮”。
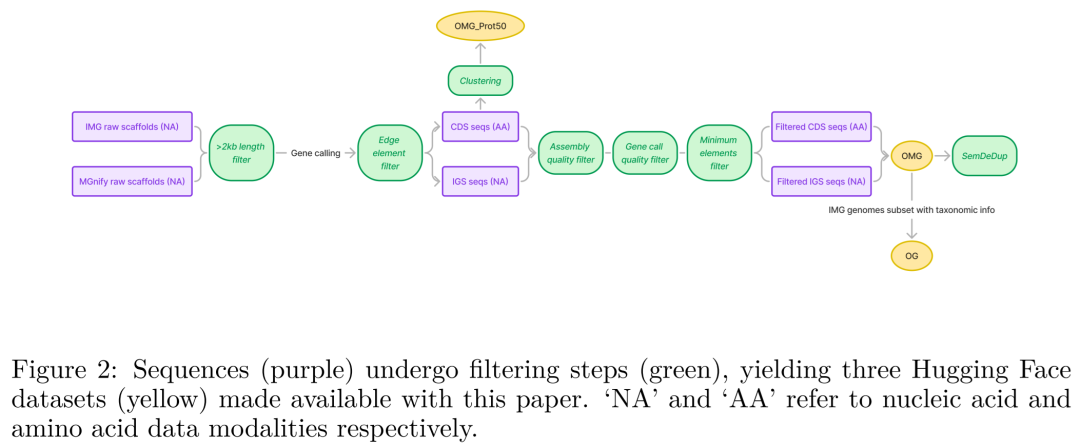
为了解决这一痛点,本文作者团队推出了Open MetaGenomic (OMG) corpus。他们整合了全球最大的两个宏基因组库(JGI的IMG和EMBL的MGnify),经过严苛的质量过滤,最终产出了一个包含3.1万亿碱基对(bp)、33亿条蛋白编码序列(CDS)的超级数据集!
为了让大家直观感受OMG的体量,我们直接看论文给出的核心数据统计表:
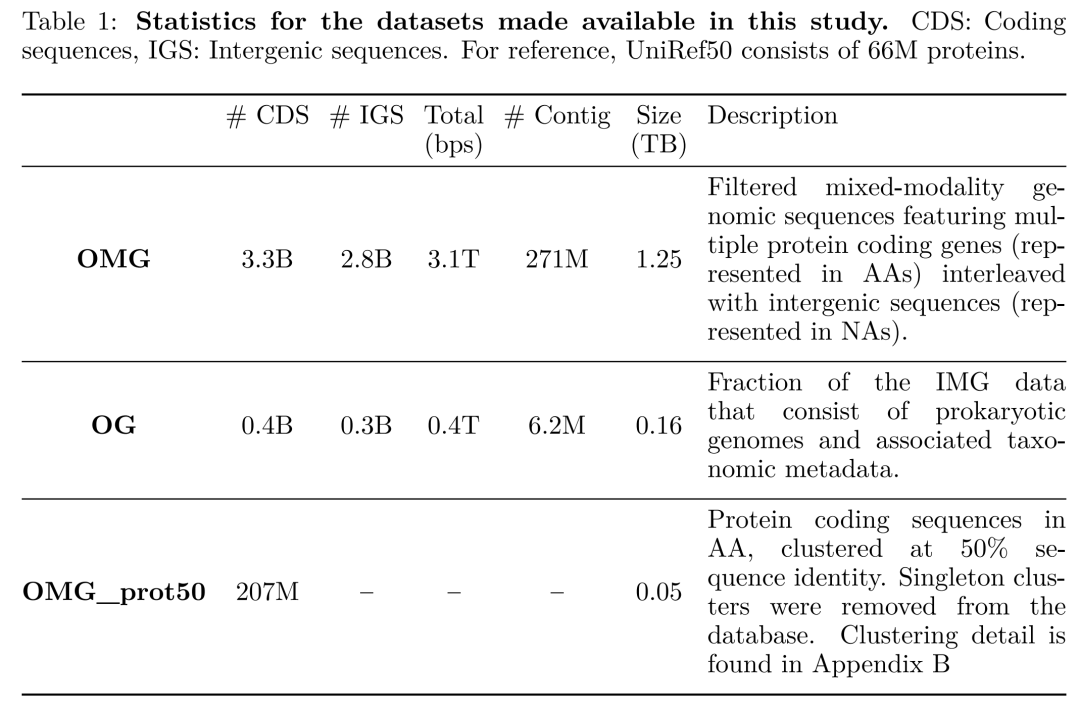
该表格展示了OMG、OG以及OMG_prot50三个数据集的核心统计指标,包括CDS数量、IGS数量、总碱基对数等,凸显了OMG比传统UniRef50大数倍的压倒性规模优势。
脑洞大开:什么是“混合模态(Mixed-Modality)”?
读到这里你可能会问:基因序列不都是用A、T、C、G表示的吗?有什么模态之分?
这就触及了本文的第一个核心创新点。传统的基因组模型通常面临两难:
纯核酸(NA)模型:直接输入ATCG。优点是能保留所有信息;缺点是序列太长,计算成本极高,且模型难以捕捉到高阶的蛋白质功能特征。
纯蛋白(AA)模型:先把基因翻译成氨基酸序列再输入。优点是压缩了长度,模型容易学到蛋白功能;缺点是丢掉了基因间区(IGS)的调控信息(比如启动子、转录因子结合位点等)。
作者的解法简单而优雅:我全都要!(Mixed-Modality)
他们将一条基因片段看作一个有序的列表:
遇到蛋白编码区(CDS),将其翻译为氨基酸(AA)输入。
遇到基因间区(IGS),保留其原始的核酸(NA)序列输入。
同时还引入了 <+>和 <->特殊Token来标记基因的正负链方向。这种设计不仅大幅度压缩了序列长度,还完美保留了非编码区的生物学意义!
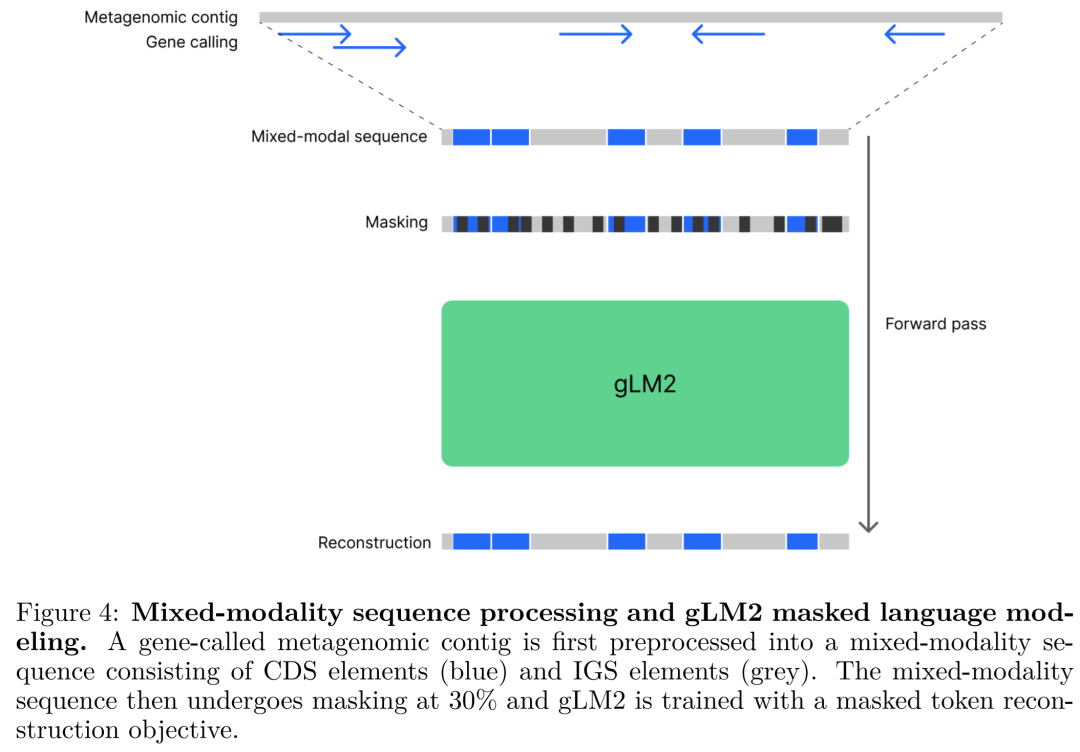
该图直观展示了如何将含有多个CDS和IGS的Contig,转化为AA和NA交错排列的混合模态序列,并附带了正负链的Token标识。
神来之笔:“语义去重”巧妙平衡数据集
如果你尝试过自己构建数据集,一定会遇到一个头疼的问题:数据偏斜(Imbalance)。比如人的肠道微生物测序很容易做,导致数据集中充斥着常见的细菌;而深海、高温泉中的罕见微生物数据极少。
传统的去重方法(如基于序列相似度的聚类)在面对长短不一的宏基因组片段时计算量极大,甚至不可行。
本文巧妙地借用了NLP领域的语义去重(Semantic Deduplication, SemDeDup)思想:
先拿一个小的gLM2模型对全量数据进行嵌入(Embedding)。
在隐空间(Latent Space)计算余弦距离。
剔除那些挤在一起的、语义相似的冗余样本。
结果令人惊喜!在不依赖任何外部分类学标签的情况下,这种基于嵌入空间的去重不仅剔除了冗余,还自然而然地实现了数据集的物种平衡,让模型对罕见物种的下游任务表现大幅提升!
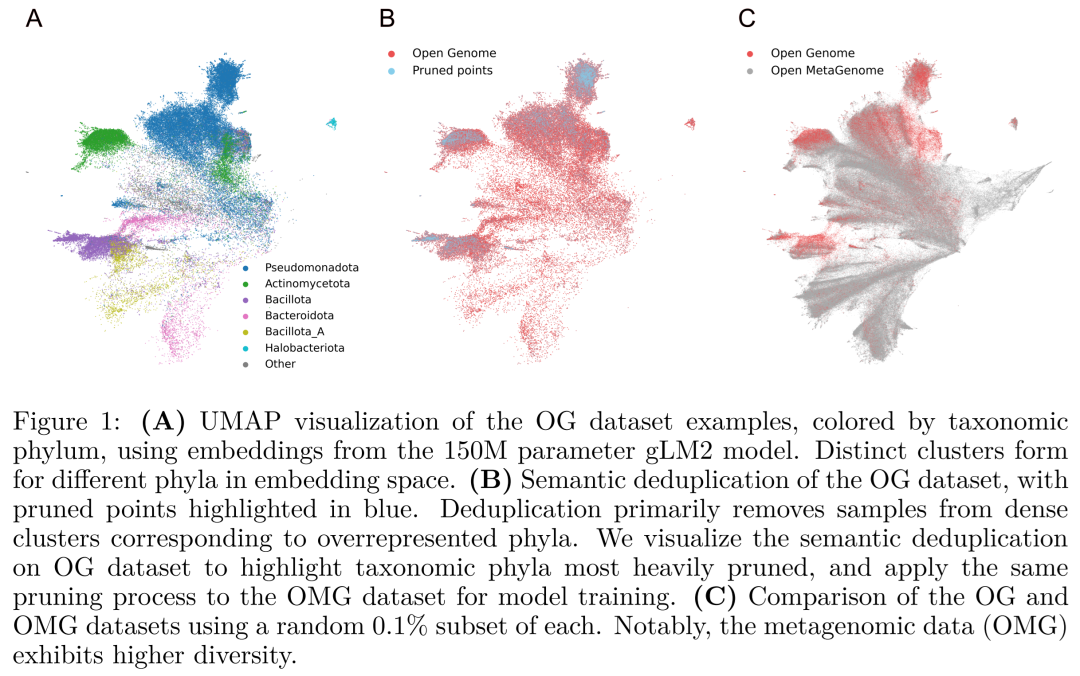
该图展示了去重前后数据集在嵌入空间中的分布变化,直观呈现了去重如何打散密集簇并提升数据多样性。
降维打击:一专多能的 gLM2 模型
有了高质量的混合模态数据,作者顺势推出了基于Transformer的基因组语言模型——gLM2。它有1.5亿和6.5亿两种参数量版本,采用了2048的上下文窗口,足以覆盖多个连续基因。
一经评测,gLM2立刻展现了“六边形战士”的潜质:
1. 超越ESM2的蛋白质任务表现
在权威的DNA与基因组评估基准(DGEB)上,gLM2在绝大多数蛋白质相关任务上的表现都超越了同等规模的ESM2(基于纯蛋白训练的SOTA模型)!
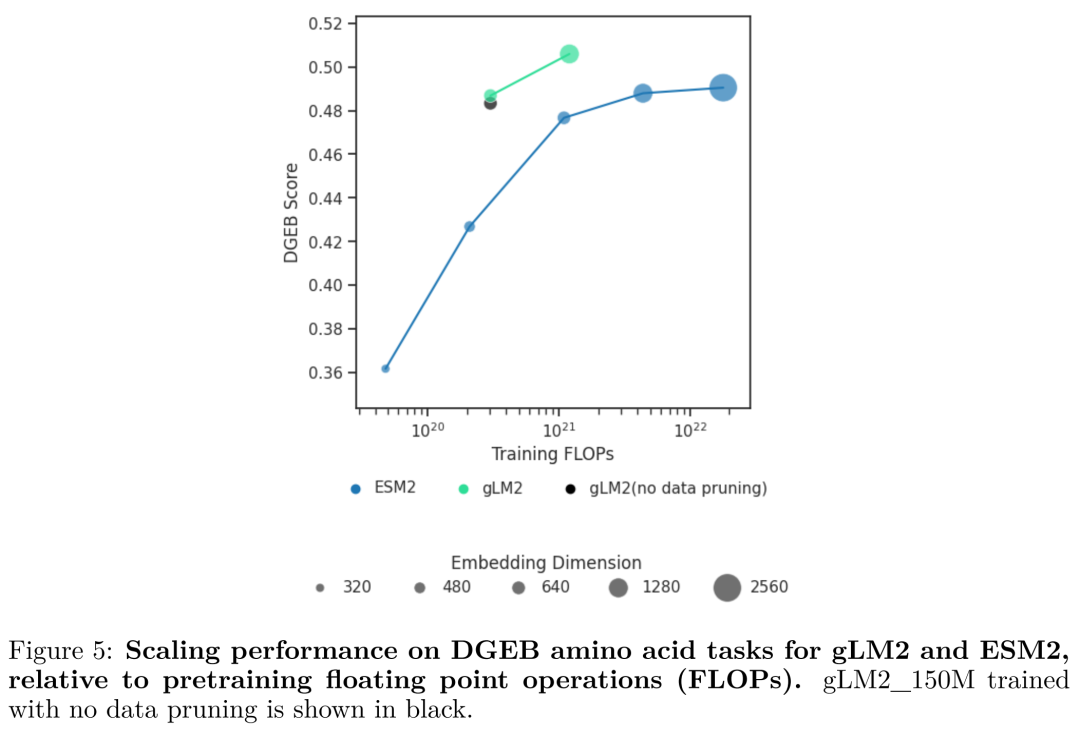
对比了gLM2与ESM2在不同任务上的表现。可以明显看出gLM2在蛋白质功能预测等任务上的面积覆盖率大于ESM2。
2. 无师自通的 PPI(蛋白质-蛋白质相互作用)预测
这是gLM2最惊艳的能力。因为传统的蛋白模型(如ESM)通常单独处理一个蛋白,而gLM2在预训练时就看到了基因在基因组上的共线性(Multiple genes in a row)。
得益于这种多基因上下文信息,gLM2天然习得了协同进化信号(Coevolutionary signals)——即当两个蛋白发生物理相互作用时,它们的基因往往相邻,且序列变异是相互制约的。实验证明,gLM2在蛋白质互作界面(Protein-Protein Interfaces)的预测能力上大幅超越了基线模型。
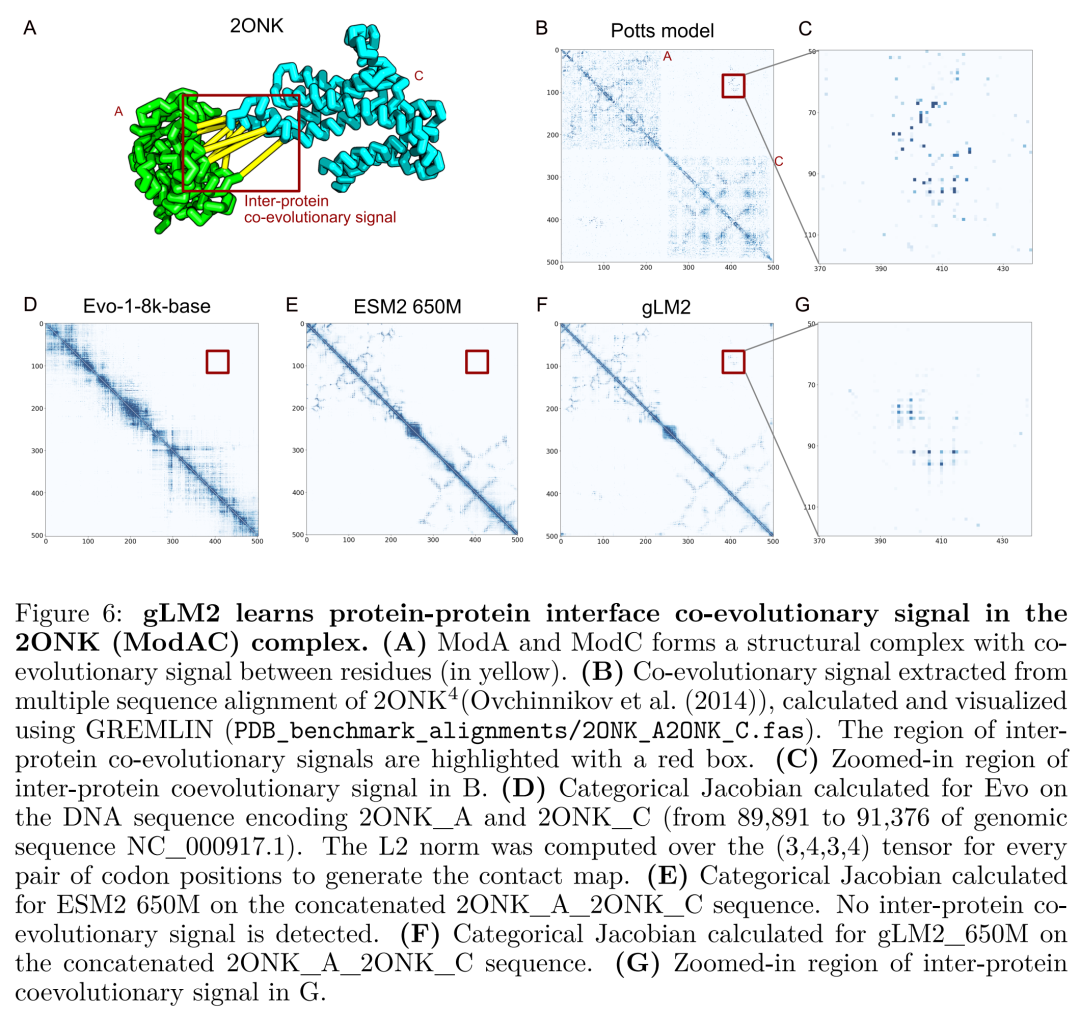
该图通过具体案例的可视化热力图,对比了gLM2预测结果与真实物理结合界面的高度重合,证明了混合模态预训练的有效性。
总结与展望
这篇ICLR 2025的论文给我们带来的不仅是庞大的数据和强悍的模型,更是一种跨学科的方法论启示:
数据层面:巧妙的“混合模态”设计和借助隐空间向量的“语义去重”,打破了生物学数据难以清洗和平衡的僵局。
模型层面:证明了引入基因组上下文(多基因共线性)可以为模型注入协同进化的先验知识,从而在蛋白质互作等关键任务上实现质的飞跃。
对于从事“AI+生物”交叉领域的同学们来说,这无疑是一个极具价值的里程碑式工作。



内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢