

文献来源:Grosjean H. & Biggin P.C. Developments and challenges in hit progression within fragment-based drug discovery.Nature Communications (2026) 17:2226.
DOI:https://doi.org/10.1038/s41467-026-68941-z
作者单位:牛津大学生物化学系结构生物信息与计算生物化学组;Diamond Light Source
目录
1. 背景与意义:为什么FBDD仍是前沿? 2. FBDD基础框架:片段的定义与筛选逻辑 3. 片段库设计:效率的源头 4. 命中物识别与验证:三类实验方法的系统比较 5. 共价片段:靶向难成药蛋白的新维度 6. 命中物选择标准:什么是"好片段"? 7. DMT循环:迭代优化的核心引擎 8. 设计阶段:从化学空间探索到AI生成分子 9. 合成阶段:高通量化学的崛起 10. 测试阶段:纯化无关方法与DEL协同 11. 整合案例分析 12. 当前挑战与未来展望 13. 总结
1. 背景与意义
1.1 Eroom定律:制药业的效率危机
过去60年,制药行业研发效率持续下滑。新药每十亿美元获批数量约每九年减半,这一趋势被称为 Eroom定律(Moore定律的反面)。这种效率衰退与治疗需求的持续扩张(老龄化、新发传染病、耐药性问题)之间的矛盾,构成了推动FBDD发展的根本动力。
1.2 FBDD的成就与地位
片段药物发现(Fragment-Based Drug Discovery, FBDD)已发展为现代小分子药物发现的主流范式之一。目前已有 6个FDA批准药物 直接源自FBDD,代表性品种包括:
此外,FBDD的应用边界已超越传统小分子,延伸至:
• 蛋白-蛋白相互作用(PPI)抑制剂的开发 • PROTAC(靶向蛋白降解嵌合体)中E3连接酶配体的发现 • 分子胶(molecular glues)的设计 • 蛋白运动探针、结合位点发现、成药性评估等生物学工具
1.3 本文的核心视角
本综述不同于以往FBDD综述对筛选方法、靶点或适应症的单一聚焦,而是以DMT(Design-Make-Test)循环为主轴,系统阐述片段命中物从初始弱亲和力结合到临床前先导化合物这一过程中的方法进展与核心挑战。作者的背景涵盖计算辅助配体设计、X射线晶体学、生物物理学和direct-to-biology测试技术,使本文具有独特的整合视角。
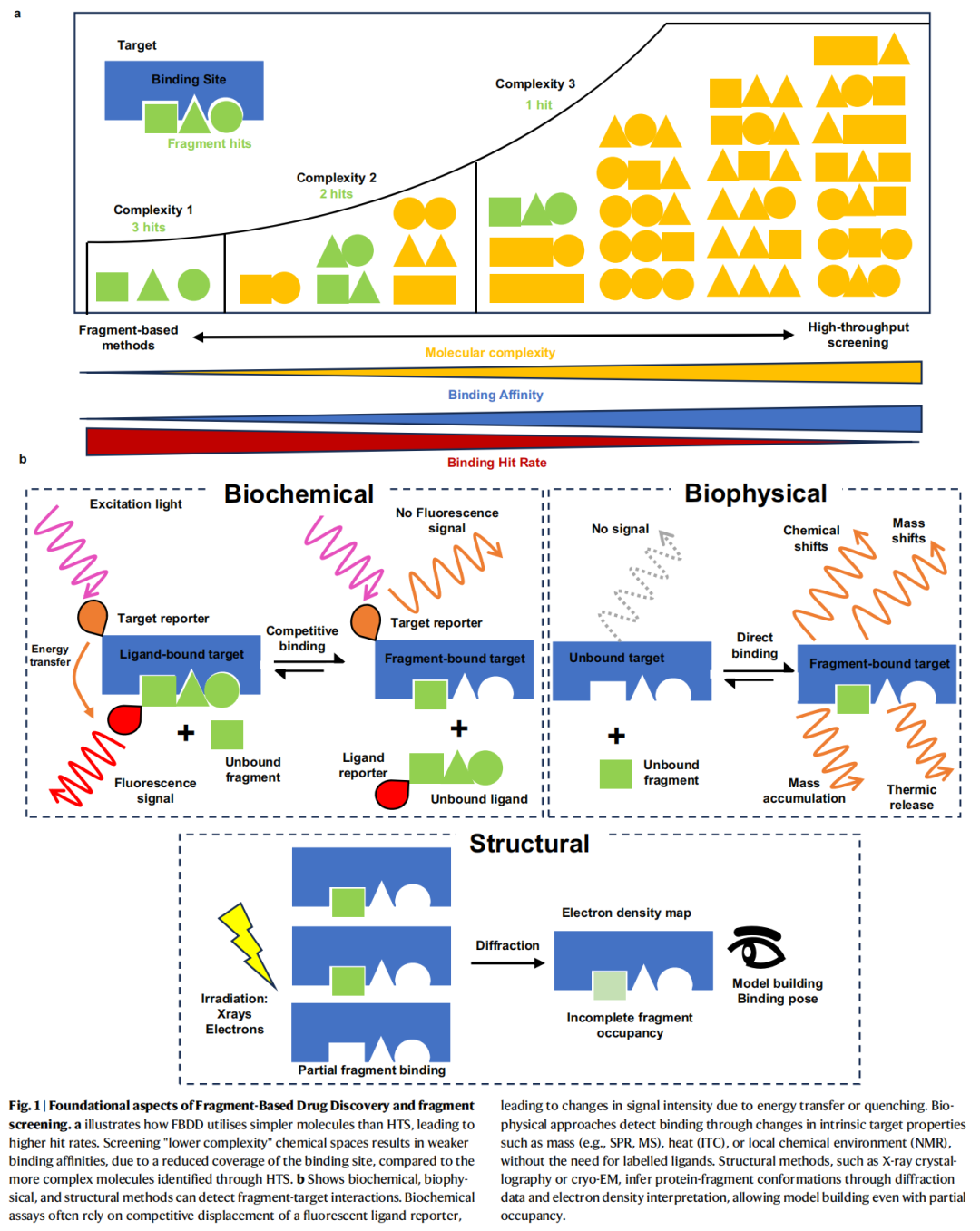
2. FBDD基础框架
2.1 片段的定义:"三规则"
片段通常由"Rule of 3"定义:
这一定义使片段处于化学空间的"低复杂度"区域,与HTS中典型的先导化合物(MW ~400–500 Da)形成鲜明对比。
2.2 FBDD vs HTS:核心差异
HTS FBDD
─────────────────────────────────────────────────────
库容量:> 100万化合物 库容量:数百至数千片段
命中率:< 0.1% 命中率:1–10%(更高)
亲和力:纳摩尔级命中 亲和力:毫摩尔至微摩尔
分子量:~400–600 Da 分子量:< 300 Da
配体效率(LE):往往偏低 配体效率:较高(≥ 0.3 kcal/mol/HA)
优化空间:受限于复杂骨架 优化空间:模块化,可创意扩展2.3 配体效率(LE):片段优先级的核心指标
配体效率(Ligand Efficiency)定义为:
LE ≥ 0.3 kcal/mol/HA 通常被认为是理想片段的基准。另一常用指标是亲脂性配体效率(LLE),表示在不依赖过度疏水性的前提下所获得的结合效率,LLE = pIC₅₀ − logP,其值越高,说明化合物以极性相互作用驱动结合,这在后续优化中更有潜力维持良好的ADMET性质。
3. 片段库设计
3.1 库设计的核心原则
片段库的质量直接决定了后续DMT循环的效率。优秀的片段库需满足:
多样性维度:
• 药效团多样性:覆盖氢键供体/受体、芳环、带电基团等不同相互作用类型的组合 • 形状多样性:平面vs三维、刚性vs柔性骨架 • 化学空间多样性:最小化功能冗余,最大化结合位点探索密度
合成可及性:
• "Synthetically Sociable":有明确的生长向量,可通过高产率、广适性的反应进行快速扩展 • 可以解构(deconstruct)以分析关键亚结构的SAR贡献
质量控制:
• DMSO溶解度 > 10 mM(确保高浓度筛选) • 化学稳定性(避免降解/聚集) • PAINS/毒性基团过滤
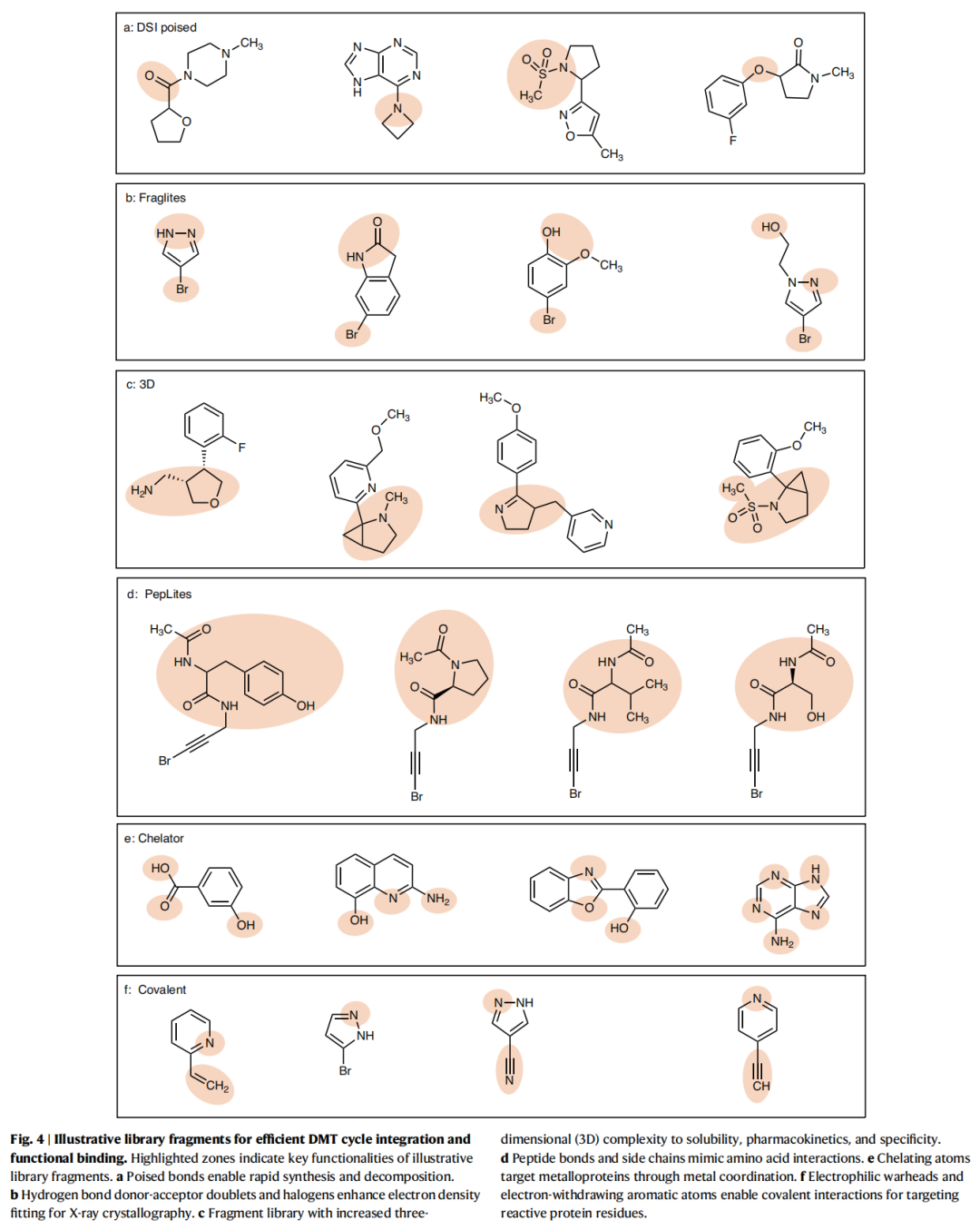
3.2 专业化片段库类型与代表案例
| Poised库 | |||
| 卤代探针库 | |||
| 三维富集库 | |||
| 拟肽库 | |||
| 螯合库 | |||
| 共价库 |
DSI-poised库案例深析:
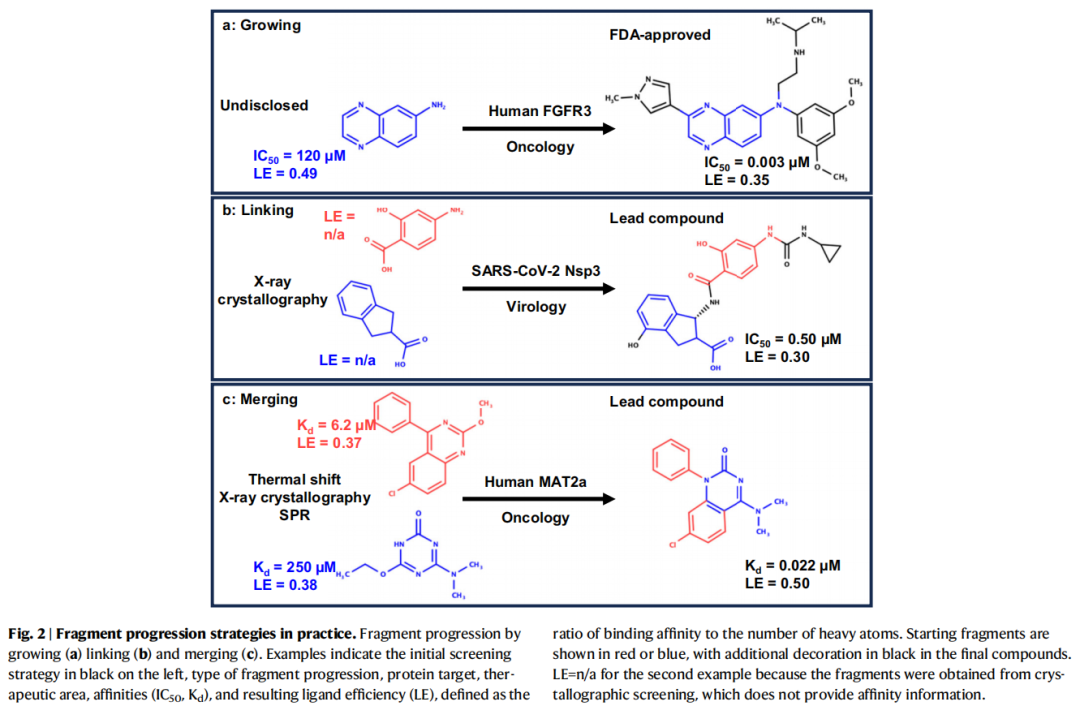
该库被用于靶向PHIP2(pleckstrin同源结构域相互作用蛋白第二溴域),一种在致命癌症中过表达的蛋白。"活化键"策略允许在保持与受体关键相互作用的同时,快速通过高产率反应扩展化学空间,最终获得首批具有可测IC₅₀值、良好配体效率和结构数据的化合物。
4. 命中物识别与验证
4.1 三类实验方法的系统比较
FBDD中片段命中物的实验检测方法可分为三大类,各有其适用场景、灵敏度与通量权衡:
4.1.1 结构方法(Structural Methods)
X射线晶体学(XRC)是FBDD的"黄金标准"结构方法:
• 通过晶体浸泡(soaking)实现高通量筛选,单周期可处理100–1000个样品 • 灵敏度可达毫摩尔级(因高浓度浸泡) • 利用PanDDA(Pan-Dataset Density Analysis)等多数据集方法提取低占位(低信号)结合事件 • 关键限制:晶体堆积可能阻止溶剂通道访问;结晶条件可能非生理构象;DMSO等溶剂可竞争结合位点(尤其溴域蛋白)
重要警示:X射线晶体学与溶液相方法(SPR、NMR等)之间的命中物重叠率通常较低。晶体学命中物在溶液检测中可能无信号,原因包括:①高浸泡浓度超出溶液相方法的灵敏度范围;②不同实验条件(缓冲液、固定化方式)影响结合检测;③晶格约束稳定非优势结合构象。这一现象具有重要实践意义,不代表假阳性,而是提示方法互补性。
冷冻电镜(Cryo-EM)在FBDD中的应用正在兴起:
• 适合生理相关构象研究,特别是膜蛋白等难成药靶点 • 目前可靠分辨配体姿态通常需要局部分辨率 ≲ 2.5–3.0 Å • 通量仍低(10–50个样品/周),尚不适合大规模初步筛选 • 与X射线晶体学的分辨率定义不同(X射线:衍射极限;Cryo-EM:粒子图像内部一致性),比较时需谨慎
NMR结构信息(有限):
• 全三维蛋白-配体复合物结构因分子量限制和同位素标记要求,实际应用较少 • 主要通过化学位移扰动(CSP)提供残基级相互作用图谱,映射到已有晶体结构上
4.1.2 生物物理方法(Biophysical Methods)
NMR的特殊地位:NMR被广泛认为是溶液相小到中等分子量靶点片段结合检测的黄金标准。配体检测NMR(STD-NMR、WaterLOGSY等)假阳性率低,可同时处理混合样品(通量友好);蛋白检测NMR(¹H-¹⁵N HSQC)则可同时提供结合位点信息,为准结构信息来源。
4.1.3 生化方法(Biochemical Methods)
包括酶活性测定、竞争性荧光偏振(FP)、时间分辨荧光共振能量转移(TR-FRET)、AlphaScreen等。通量最高(1000–10000/天),但对片段更易受PAINS干扰,假阳性率相对较高,通常作为初步筛选使用,需正交方法验证。
4.2 PAINS:假阳性的系统性陷阱
**泛实验干扰化合物(PAINS)**是FBDD中特别需要警惕的问题。由于片段在检测极限附近工作,PAINS通过以下机制产生靶标无关信号:
• 胶体聚集:在高浓度筛选条件下形成纳米颗粒,非特异性捕获蛋白 • 氧化还原循环:如醌类、罗丹宁类化合物,干扰基于氧化还原的酶活检测 • 共价反应性:Michael受体等与蛋白非特异性修饰 • 金属螯合:螯合活性中心金属,产生假性抑制 • 荧光干扰:内滤效应(inner filter effect)或荧光淬灭/增强
典型PAINS结构特征:儿茶酚/醌、罗丹宁、偶氮染料及相关氧化还原活性骨架。
结构方法的内在优势:晶体学或其他结构方法本质上减少了PAINS问题,因为其结合证据来自物理合理的姿态(清晰的差异电子密度、合理的几何形状和相互作用模式),而非间接的信号变化。
4.3 正交验证策略
多方法正交验证是片段命中物质量控制的核心原则,理想的验证级联为:
初步筛选(高通量生化/生物物理)
↓
正交验证(不同原理的第二种方法)
↓
结构确认(晶体学/NMR,有条件时优先)
↓
功能验证(酶活性/细胞活性,排除结构沉默结合)跨独立方法的一致读出提供更强的真实靶标结合证据,支持优先级排序。
5. 共价片段
5.1 共价策略的原理与优势
共价片段携带能与蛋白亲核残基不可逆反应的**"弹头"(warhead)**。共价结合通过将片段"钉住"在反应性残基上,从根本上规避了FBDD的核心挑战——弱亲和力问题。其优势包括:
• 信号放大:质谱(MS)通过蛋白质量变化直接读出共价加合物,无需高灵敏度 • 特异性:弹头经过调校,足够反应活性以靶向特定残基,但不随意与非靶点亲核基团反应 • 难成药靶点:适合浅表或动态结合口袋("undruggable"蛋白)
5.2 弹头化学的进展
磺酰氟是近年热点:能靶向多种亲核残基,但需注意其在DMSO或水中可能水解,同时也是组合化学中有用的构建块。
5.3 共价筛选验证的特殊挑战
共价结合违反经典平衡假设,标准动力学分析(如SPR直接读出Kd)不再适用,需要专门的验证流程:
• 定量不可逆拴系(qIT):比较片段对靶蛋白与非特异性硫醇(谷胱甘肽)的反应动力学,区分靶向性共价与随机标记 • 时间依赖性酶抑制:酶活测定中时间依赖性特征提示共价机制 • 突变验证:靶向残基(如Cys)的点突变导致活性丧失,是最直接的特异性证据 • MS/MS:串联质谱定位修饰残基
代表性成功案例——Sulfopin(Pin1抑制剂):
通过MS筛选电亲核片段库,发现以磺内酯-氯乙酰胺弹头共价修饰Pin1的Cys113位点。Sulfopin对Pin1具有强特异性、低毒性,在小鼠肿瘤模型中展示肿瘤消退,成为Pin1靶向治疗的重要探针化合物。
5.4 共价FBDD的ADMET考量
不可逆共价结合引入独特的ADMET挑战:
• 延长居留时间:可能导致剂量优化复杂化 • 蛋白降解/免疫清除:共价加合物蛋白可能被泛素化降解或引发免疫反应 • 合成中弹头需在较晚阶段引入,以避免与合成中间体发生副反应
6. 命中物选择标准
6.1 关键评估维度
片段命中率通常高于HTS,选择哪些片段进行后续发展是决定项目成功的关键决策。综合文献与作者经验,优质片段应满足以下多维度标准:
6.2 结合热力学的深层含义
焓驱动结合(通过定向极性接触和去溶剂化)通常预示更高的靶标特异性和结合稳定性。相比之下,单纯由疏水相互作用(熵驱动)贡献的结合在先导化合物优化中往往需要引入更多脂溶性基团,带来ADMET风险。因此,具有良好焓贡献的片段是更理想的起始点。
6.3 结合位点的生物学相关性
结合"热点"(hotspot)——蛋白能量学上重要的结合区域——是片段优先化的关键考量:
• 功能性(催化位点、变构位点)优于溶剂暴露的外围位点 • 对于激酶等高度保守靶点家族,变构位点结合片段具有更高选择性潜力 • "沉默结合者"(Silent Binders):在热点以外结合的片段,生物物理阳性但无生化活性。虽对直接活性优化无意义,但可作为PROTAC配体候选(利用其结合靶蛋白的能力)
7. DMT循环
7.1 循环框架与迭代逻辑
设计-合成-测试(DMT)循环是FBDD命中物优化的核心组织框架。本质上是一个数据驱动的迭代优化系统:
片段命中物
│
▼
┌─────────────────────────────────┐
│ 设计(Design) │
│ 配体法 / 结构法 / AI/ML辅助 │
│ SAR分析 → 合成可行性评估 │
└───────────────┬─────────────────┘
│ 设计方案(周至天)
▼
┌─────────────────────────────────┐
│ 合成(Make) │
│ 路线规划 → 合成执行 → 质控 │
│ 纯化 或 纯化无关策略 │
└───────────────┬─────────────────┘
│ 化合物(天至周)
▼
┌─────────────────────────────────┐
│ 测试(Test) │
│ 结合活性 / 结合姿态 / ADMET │
│ → 新SAR数据 │
└───────────────┬─────────────────┘
│ 数据反馈(周至月)
▼
先导化合物系列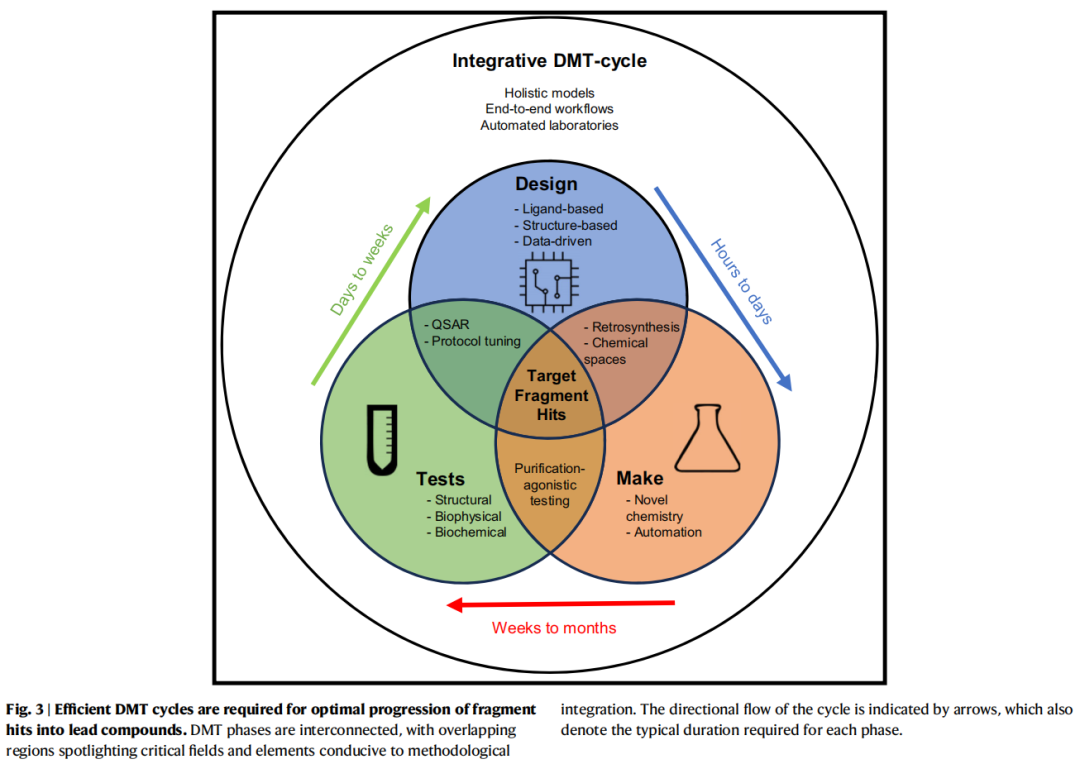
7.2 早期阶段:SAR基础建立
在DMT循环的早期阶段,核心目标是围绕片段命中物建立可靠的SAR模型:
"目录SAR"(SAR-by-Catalogue)策略:筛选商业可得类似物,以低成本快速探索命中物周边化学空间。该策略的优势:
• 无需定制合成,速度快、成本低 • 支持自动化(结合晶体学结构可实现全流程自动化) • 为深入定制合成提供优先化依据
代表性工具流程——Frag4Lead:利用分子对接虚拟筛选MolPort目录,从超过10,000个候选分子中筛选endothiapepsin的5个片段命中物的28个扩展化合物,10个经晶体学确认,5个显示亲和力提升。
7.3 进阶阶段:先导系列鉴定与多参数优化
随着DMT循环推进,目标从SAR数据获取转向化合物效力的系统提升(趋向纳摩尔级亲和力):
• 平行追踪多个化学系列(备份策略),防止单一系列遭遇瓶颈 • 引入早期ADMET测定:代谢稳定性(CYP P450,MDR1外排)、hERG心脏毒性筛选、细胞渗透性 • 精细化学修饰(如特定立体化学、空间位阻控制)对合成要求更高 • 在此阶段,**计算自由能方法(FEP)**可辅助优化,但前提是有可靠的配体结合姿态
7.4 FBDD-DMT vs 传统药化优化的本质差异
7.5 协同案例:COVID Moonshot与开放科学范式
COVID Moonshot针对SARS-CoV-2主蛋白酶(Mpro)的全开放科学项目,是迄今FBDD领域规模最大的协同DMT实践:
• 筛选规模:晶体学片段筛选发现71个命中物(包括共价结合物),位于催化活性位点和二聚体界面 • 全球协作:众包设计超过18,000个化合物提案,2400个实际合成,490个配体-蛋白X射线结构 • 技术整合:机器学习辅助合成 + 分布式测试 + 自动化结构分析 • 最终产出:预临床候选化合物ASAP-0017445,设计为"直接仿制药"以促进全球可及性
该项目证明,在资源约束下,开放科学与自动化的结合可以实现令人瞩目的DMT效率。
8. 设计阶段
8.1 计算层级:从配体法到自由能计算
设计阶段的计算方法构成一个由粗到精的层级体系,计算成本与预测精度呈正相关:
计算成本递增 ──────────────────────────────────→
配体法(2D/3D)→ 分子对接 → 分子动力学 → 自由能计算(FEP)
处理化合物数量递减 ←──────────────────────────────
预测精度递增 ──────────────────────────────────→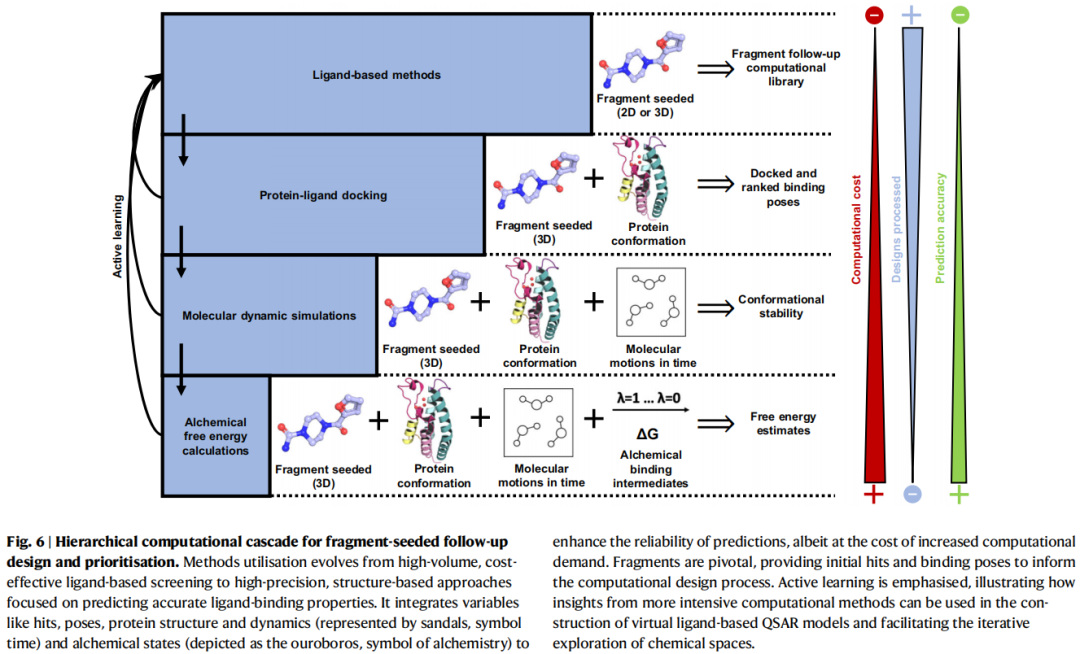
8.2 化学空间探索与配体法
建立初始跟进化学空间:
• Enamine REAL Space(2025版):78亿个可合成类药分子,通过验证反应规则和建筑块枚举,交付周期3–4周,成功率约80% • Fragment Network(图数据库):将片段连接到商业可得类似物网络,支持片段融合搜索,已在Mpro和结核分枝杆菌EthR靶点中回顾性验证 • Syndirella框架:合成导向的片段扩展,基于反应规划生成每个骨架数百到数千个同系物,报告成本较目录购买降低约80%
生成模型与变分自编码器(VAE):
• Fragment-based Sequential Translation(FaST):通过VAE学习片段分布,以添加/删除操作导航化学空间,多目标优化(生物活性、新颖性、合成可行性) • 配体基生成模型可以片段骨架为种子,约束搜索空间并同时优化多个参数
ADMET早期整合:临床衰减约40–45%归因于ADMET问题。基于逆合成路径的机器学习模型(CoPriNet等)可预测化合物价格或合成成本,支持早期优先化。
8.3 结构导向对接:片段姿态的整合
核心挑战:常规对接工作流程未内嵌对实验片段姿态的利用,需要量身定制协议:
1. 片段姿态约束对接:锁定已知的片段结合几何,在此基础上生成先导化合物姿态 2. 形状/药效团叠合评分(SuCOS等):先导化合物与原始片段的三维相似性评分,优选保留关键相互作用的姿态 3. 电子密度直接利用:在已有实验电子密度的情况下,引导对接姿态与真实密度吻合
代表性成果:以4个PKA(蛋白激酶A)片段姿态为模板进行模板对接虚拟筛选,从中发现40个活性化合物,最优跟进化合物较原始片段亲和力提升13,500倍。
共价片段的特殊处理:标准对接工具不适用于共价结合,需要专门的共价对接协议(如Glide、AutoDock4/GPU版本中的共价模式),以弹头-残基共价键为约束进行采样。
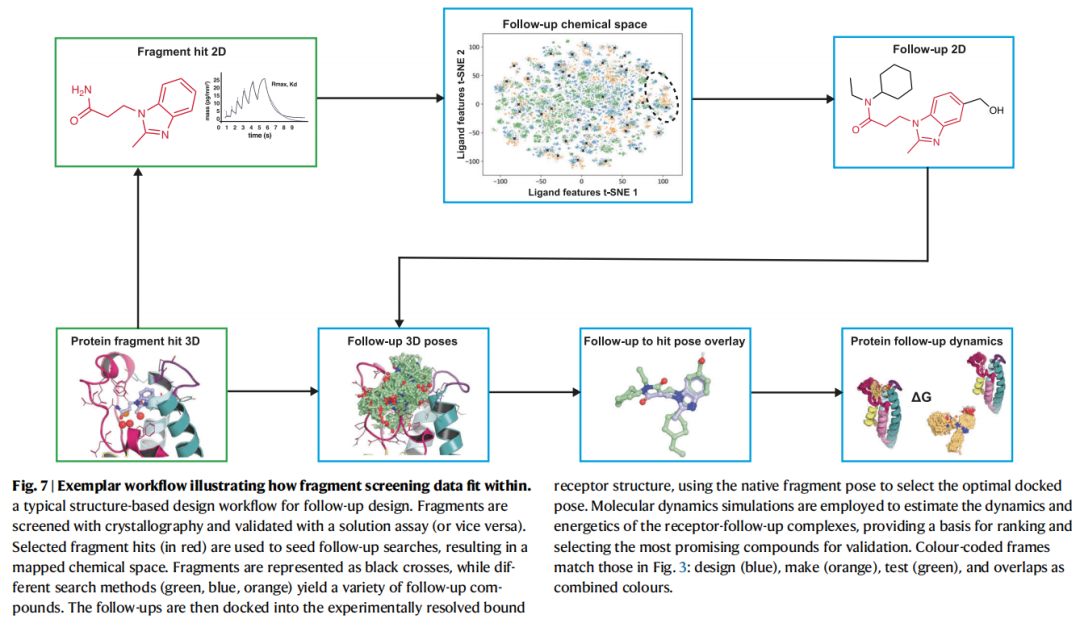
8.4 分子动力学(MD)在设计阶段的应用
用途分层:
关键案例——A1腺苷受体(A1AR):NMR筛选得到微摩尔级片段后,通过MD和RBFE计算预测一系列扩展化合物的结合自由能,实验结果显示最优化合物亲和力提升41,000倍,选择性提升40倍。
片段连接热力学分析(Yu et al., 2021):通过自由能分解分析,揭示片段连接中预期的亲和力加和性(additivity)常被破坏的原因——连接臂引入的不利相互作用、结合模式改变以及熵损失抵消了潜在收益。这为"片段连接为什么经常失败"提供了定量框架。
8.5 主动学习结合结构方法
主动学习(Active Learning)策略的核心思路:用少量高精度(但昂贵)的结构计算结果训练快速配体模型,然后用此模型快速评分大规模化学空间:
• Thompson采样 + 形状筛选:从22亿分子的Enamine REAL库中,以SARS-CoV-2大结构域的两个结晶片段命中物为参考形状,仅用极小比例的计算量完成全库虚拟筛选,发现低微摩尔级片段融合物 • Link-INVENT:结合强化学习与对接评分,优化片段连接臂的生成 • FEP + 主动学习:以FEP评估少量化合物后训练ML模型,迭代发现高亲和力PDE2抑制剂
8.6 生成式AI的前沿与局限
三维分子生成模型:
当前核心问题(作者直言不讳地指出):
1. 当前de novo三维生成方法倾向于产生化学不可行结构和物理上不合理的结合姿态 2. 数据稀缺性(相对化学空间)严重限制模型泛化能力 3. 可合成性、可解释性和可靠验证仍是集成到常规流程的主要障碍 4. 协同折叠(co-folding)方法目前在泛化性方面表现不佳
9. 合成阶段
9.1 合成阶段的特殊挑战
在FBDD-DMT中,合成阶段往往是限速步骤:
• 未预期的副反应、低产率、劳动密集型纯化 • 化合物格式化(溶剂、浓度兼容性)占用大量时间 • 工业实验室倾向成熟高产率策略;学术和早期阶段公司更多采用新兴方法
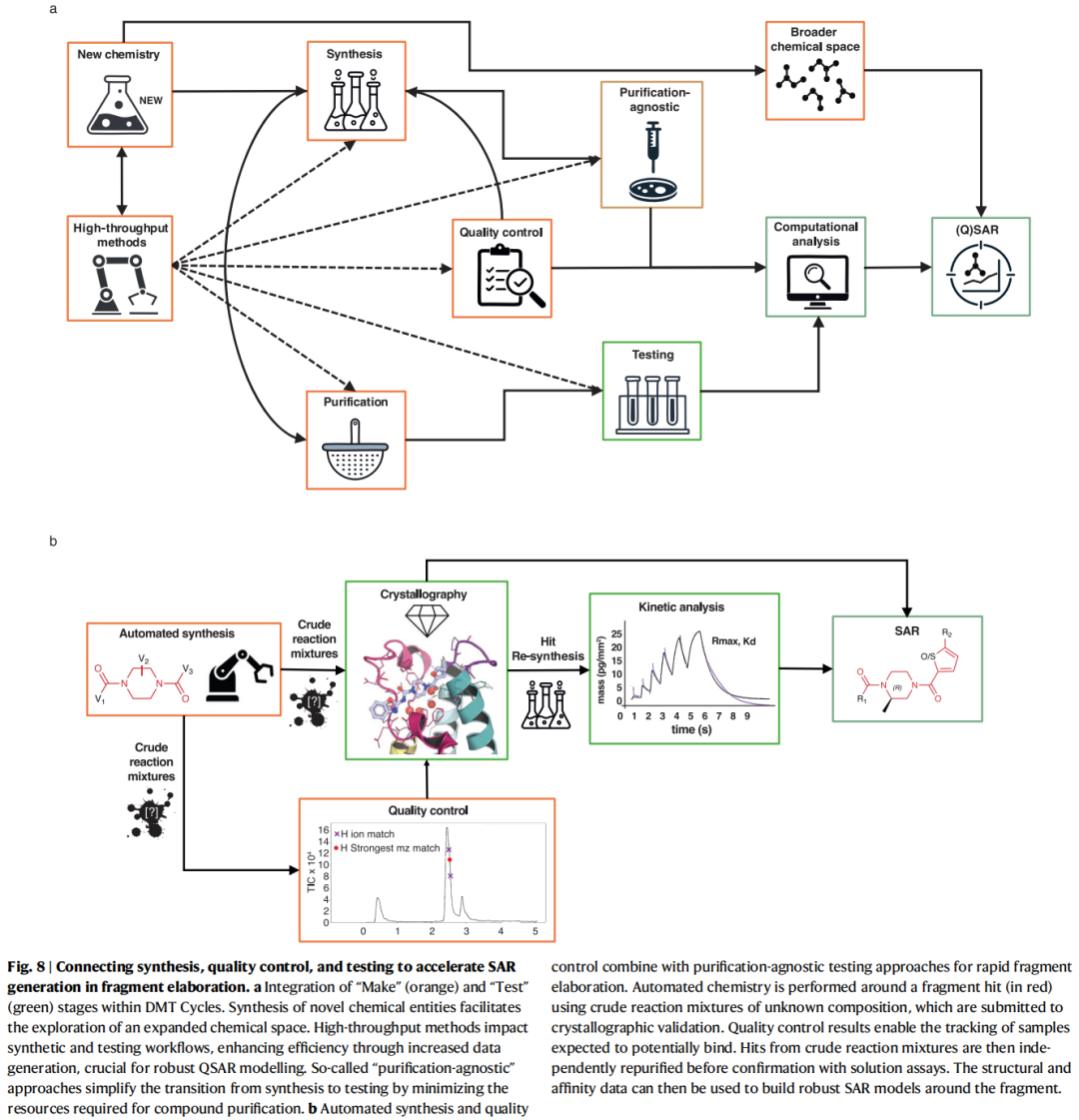
9.2 计算辅助合成规划
逆合成算法的作用:将设计分子解构为简单组分,映射最优合成路径。
• 以片段或其类似物为约束起始原料,生成符合特定结构要求的合成方案 • 代表平台:SPAYA™(Iktos,可商业获得),支持将片段命中物或类似物设为约束原料 • 几何深度学习 + 高通量反应筛选(Nippa et al., 2023):预测反应产率和区域选择性,识别小分子结构多样化机会
9.3 高通量实验平台
声波液滴喷射(ADE)技术:
• 384孔板并行合成,围绕16种不同类片段骨架生成大规模多样化合物库 • 代表了"化学速度"——用自动化实现高通量
簇合成策略(Cluster Synthesis):
• 将化学多样的反应按操作窗口(温度/时间重叠)批量排程,在单台机器人上执行 • 27种命名反应、135个分子,在三次实验中完成 • 大幅提升结构多样性和平台利用率
流动化学(Flow Synthesis):
• 多达8种合成方法集成的连续流平台,产出速率可达4个化合物/小时 • 整合金属光氧化还原偶联等新型化学 • 与高通量纯化系统配合,实现化学多样片段衍生物的全自动化生产
端到端自动化平台(Abdiaj et al., 2023):
• 集成了液-液萃取和质控,无人值守合成和纯化 • 采用Negishi偶联反应,产物富含sp³杂化碳(有利于成药性) • 减少了纯化这一传统DMT瓶颈
9.4 关键合成方法进展
C-H活化在FBDD中的革命性作用(Chessari et al., 2021):
• 约80%的片段生长来自碳中心的扩展向量,而与受体形成极性相互作用的基团往往不参与反应 • C-H活化允许直接在碳骨架上引入新取代基,无需预先引入活化基团 • 代表药物——Tolinapant(ASTX660):IAP蛋白(凋亡抑制蛋白)拮抗剂。NMR筛选获得片段后,结构导向优化,在脂肪族碳位点添加简单甲基即产生60倍效力提升,突显了精确C-H官能化的价值
碳-杂原子键形成:光催化半异质亚相金属光催化(Song et al., 2025)实现了磺酰胺/氨基甲酸酯前体的位点选择性氮/氧取代,为规模化片段衍生提供了新工具。
点击化学模块化扩展:两步法(伯胺→叠氮;Cu催化叠氮-炔环加成,形成三唑连接)可在数小时内建立大规模类似物库,产物纯度足以直接用于生物测试。已在靶向KSHV病毒蛋白LANA的片段中得到验证。
可合成化学的新领域——1,1'-双环丙烯衍生物:通过Au/Ag双金属催化环丙烯基交叉偶联合成,这类高张力双环苯异构体开辟了全新化学空间,温和条件下的模块化合成和良好的官能团容忍性,使其具备FBDD应用潜力。
9.5 反应可扩展性与溶剂考量
光氧化还原介导的交叉脱氢偶联(Grainger et al., 2019):从纳克级高通量反应条件筛选出发,经连续流化学实现克级放大,产物含sp²-sp³特权结构(杂环胺类),直接与药物化学需求对接。
溶剂干扰问题:
• DMSO是常用合成和储存溶剂,但可以模拟乙酰化赖氨酸结合基序,与溴域蛋白的药效学结合位点竞争 • 因浓度远超配体(溶剂分子浓度∞ vs 配体毫摩尔级),可饱和结合位点,阻止晶体学配体结合 • 解决方案:为晶体学实验使用乙二醇等替代溶剂;溶液相实验中设置适当的溶剂基线对照
10. 测试阶段
10.1 "纯化无关"(Direct-to-Biology)方法的战略价值
传统测试流程要求纯化化合物,而纯化是DMT循环的主要时间成本之一。"纯化无关"方法直接对粗品混合物进行测试,仅在发现命中物时才回头纯化验证:
优势:大幅压缩Make-Test周期;允许更大规模探索化学空间
代价:混合物中多种组分可能贡献信号,需更复杂的数据分析和正交验证
10.2 粗品晶体学:高通量结构测试
核心流程:
1. 平行合成产生板格式阵列(粗品混合物) 2. 直接将粗品混合物浸泡到蛋白晶体 3. 高通量自动化X射线数据收集(XChem @ Diamond Light Source) 4. LC-MS质控:追踪哪些孔含有目标产物 5. 将结构数据与质控信息交叉,提取SAR模型
B-SPA流程(Binding-Site Purification of Actives)(Grosjean et al., 2025,本文作者工作):
• 利用OpenTrons液体分配机器人围绕PHIP2片段命中物生成数百个衍生化合物 • 将粗品阵列直接浸泡到蛋白晶体,收集数百个高通量晶体学数据集 • 表明即使是粗品实验数据中存在"噪音",也可通过识别保守化学特征提取可操作的SAR模型 • 进一步展示如何以此SAR驱动商业化合物虚拟筛选,进一步加速DMT循环
粗品晶体学的局限:
• 可能解析出低亲和力组分(较预期命中物弱) • 多组分竞争进入晶体格子,产生部分占位和叠加电子密度 • 不稳定中间体或降解产物可能误导结构解释(Cramer et al.案例:降解产物干扰解析) • 必须配合严格的质控,并与溶液相正交方法联合验证
10.3 k_off(解离速率)筛选:浓度无关的SAR代理指标
原理:解离速率(k_off,1/s)仅取决于配体-蛋白相互作用的强度,不受浓度变化影响。粗品混合物浓度通常未知或不确定,因此k_off是浓度无关的结合亲和力代理指标:
SPR k_off筛选:针对BRD3-ET(溴域和额外末端结构域3 ET区域)的实例,通过NMR初步鉴定片段后,购买类似物进行NMR验证,然后对粗品平行合成混合物实施k_off筛选,发现约30倍亲和力提升的命中物,纯化后证实k_off与粗品测定一致,验证了方法可靠性。
GCI(光栅耦合干涉):对快速解离速率(短居留时间)的分辨能力优于标准SPR,适合早期片段追踪中的快k_off化合物。
10.4 NMR用于复杂混合物测试
配体检测NMR(STD-NMR、WaterLOGSY等):
• Wu et al.(2013):超过10,000个化合物以混合物格式筛选,可靠检测弱结合物,纯化后单体确认 • 对假阳性(PAINS)具有天然抵抗力(信号机制与常见PAINS干扰机制不同)
蛋白检测NMR用于组合库混合物(Alboreggia et al.,2023):
• 监测靶蛋白hMcl-1脂肪族区域的叠加化学位移扰动(CSP) • 对约125,000个化合物的焦点组合库进行混合物筛选 • 通过累积CSP图案识别共享骨架的活性基序,无需逐一纯化
NMR for SAR方法(Larda et al.,2023):
• 整合¹⁹F-NMR(信号形状变化)、弛豫型结合测定(T₁ρ、T₂)以及蛋白/配体双侧NMR • 监测溶解度、聚集、靶标稳定性和相互作用的完整图谱 • 应用于HRas等难成药靶点的命中物到先导化合物开发
10.5 动态组合化学(DCC)与蛋白导向组装
原理:片段构建块在热力学控制下可逆组装,靶蛋白将平衡推向亲和力最高的组合体,LC-MS鉴别富集产物。
优势:无需独立纯化、亲和力定量或合成所有可能组合体;纳摩尔级抑制剂可无需合成中间体直接发现。
代表案例:酰基腙片段在α-葡萄糖苷酶存在下动态组装,LC-MS比对蛋白模板库与空白库,鉴别富集产物,酶抑制验证,迭代生长/库演化获得纳摩尔级抑制剂。
10.6 DNA编码化合物库(DEL)与FBDD的协同
DEL将数百万至数十亿化合物编码在DNA标签上,亲和力选择后通过测序解码命中物,本质上是"纯化无关"的超大规模平行筛选。
与FBDD的整合策略:
1. 片段→DEL种子:X射线晶体学鉴别BRD4片段命中物(模拟乙酰化赖氨酸),以片段骨架(异噁唑弹头)为核心,通过片段-DNA偶联Suzuki-Miyaura交叉偶联,构建42成员的"活化"DEL(NUDEL)。筛选得到氨基酸样侧链 + 杂芳基帽的最优组合,最终获得51 nM BRD4抑制剂,具备良好ADMET特征。 2. DEL数据去噪ML模型:DEL读出固有噪声(不完全合成、DNA标签错误编码、测序误差等),不确定性感知ML模型(Lim et al.)可降噪处理,提升结合亲和力预测可靠性 3. Graph Neural Network + 自注意力(GNN + Attention):将DEL结合数据与对接姿态整合,进一步修正结构虚拟筛选预测(Shmilovich et al.,DEL-Dock框架)
10.7 亲和力色谱-质谱(AC-MS)
适用于复杂混合物中的结合相互作用精确定量,在平衡条件下运行,理论上不受浓度不确定性影响,是粗品混合物测试中提取定量亲和力信息的新兴工具。
10.8 从靶标结合到细胞活性:暴露差距
蛋白层面的亲和力和效力不能保证细胞活性,常见失活原因包括:
• 细胞渗透性不足:影响细胞内靶标的化合物 • 主动外排(P-gp、MRP):影响CNS和肠道渗透 • pKa/logD非优化:影响溶解度和离子化状态 • 非特异性细胞内结合:减少游离浓度
典型案例——DNA旋转酶GyrB抑制剂(AZD5099):NMR筛选得到毫摩尔级吡咯命中物,晶体学引导优化至微摩尔级ATPase抑制剂,但早期先导化合物缺乏抗菌细胞活性,揭示了酶与细胞之间的"暴露差距"。最终通过3-哌啶取代基变化和4-噻唑取代基(形成七元分子内氢键,调节酸性和渗透性)解决了外排和渗透性问题,获得临床候选化合物。这是"结构正确但细胞无效"困境的典型案例。
11. 整合案例分析
11.1 PDE10A抑制剂开发:多轮DMT循环的互补价值
靶点:磷酸二酯酶10A(PDE10A),精神科靶点(精神分裂症)
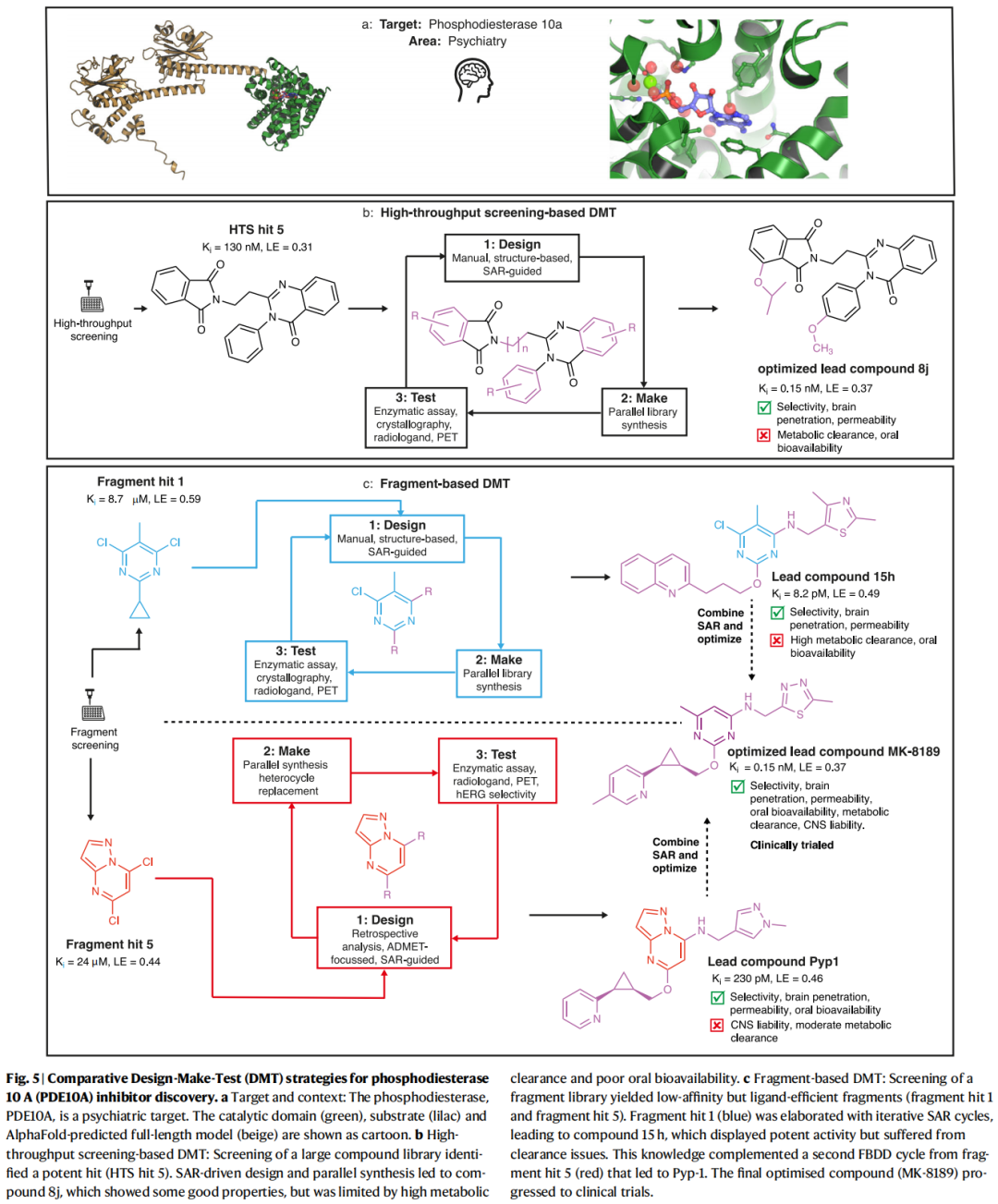
HTS路径(图5b):
• HTS命中化合物(Kᵢ = 130 nM,LE = 0.31),SAR驱动设计,平行合成到化合物8j(Kᵢ = 0.15 nM,LE = 0.37) • 优点:选择性、脑渗透性、渗透性良好 • 瓶颈:高代谢清除率,口服生物利用度差 • 8j仍作为PET探针([¹¹C]MK-8193)用于活体靶标结合评估
FBDD路径(图5c):
• 片段命中物1(Kᵢ = 8.7 μM,LE = 0.59),迭代SAR,得到先导化合物15h(Kᵢ = 8.2 pM,LE = 0.49) • 15h:选择性、脑渗透性、渗透性良好,但高代谢清除率,口服生物利用度差 • 片段命中物5(Kᵢ = 24 μM,LE = 0.44),回顾性SAR分析,ADMET导向设计,得到Pyp-1(Kᵢ = 230 pM,LE = 0.46) • Pyp-1:选择性、脑渗透性、口服生物利用度良好,但CNS毒性风险
融合优化——MK-8189:
• 整合两条路径的SAR洞察,用嘌呤衍生骨架替换初始片段的嘧啶核心,解决CNS毒性问题 • 最终:Kᵢ = 0.15 nM,LE = 0.37,优化的CNS暴露、生物利用度和代谢稳定性 • MK-8189进入临床试验
本案例深刻揭示:多条DMT路径——即使起点不同、各有瓶颈——可以系统互补,在多参数优化中实现协同突破。
11.2 A3腺苷受体拮抗剂:FEP辅助优化
以人腺苷A3受体(hA3R)拮抗剂开发为例,展示了先进计算方法与实验的无缝集成:
• 从片段命中物出发,利用FEP和MD模拟预测修饰类似物的结合自由能 • 基于计算预测优先化25个新类似物进行定制合成 • 通过测定级联(亲和力 → 选择性 → 代谢稳定性 → 毒性),获得纳摩尔级亲和力、低毒性的候选物 • 计算准确性在实验验证中得到证实
12. 当前挑战与未来展望
12.1 核心技术挑战
片段弱亲和力的固有局限——贯穿全文的核心矛盾:
• 结构测定误差增加(低占位,信噪比下降) • SAR趋势不明显(活性差异可能落在实验误差范围内) • 计算方法(对接评分、FEP)不确定性显著增大 • MD采样需要更长时间尺度才能捕获有意义的结合/解离事件
粗品数据去噪与可靠SAR提取:
• 纯化无关方法带来速度,但需要更复杂的质控和正交验证 • 多组分混合物中的真实命中物识别仍具挑战 • "混乱"数据可能隐含有价值的构象异质性信息(Fraser的观点),但也可能误导后续设计
晶体学片段数据的标准化与共享争议:
• Jaskolski等提出多数据集片段筛选数据(如PanDDA产生的数据集)需要特别标注,防止非专家误用 • Weiss等反驳这些数据集具有重要价值,捕获了构象和组成异质性 • Fraser支持拥抱"混乱"数据以揭示隐藏的动态现象 • 争议折射出领域内保守验证与开放探索之间的核心张力
片段库选择的主观性:目前缺乏普适性的片段库设计标准和命中物选择框架,高度依赖经验判断和项目特异性。
12.2 方法论边界与机会
AI/ML在FBDD中的成熟度评估:
共价FBDD的计算鸿沟:用于模拟共价结合事件的量子力学(QM/MM)工具尚不成熟、对非专家不友好,这是一个明确的方法论缺口。
12.3 公平获取与资源挑战
高通量自动化平台(机器人合成、自动化晶体学)和先进计算基础设施(GPU集群、Anton超算)需要大量资金投入,对抗病毒、抗生素等欠资助治疗领域以及中低收入国家研究机构形成壁垒。开放科学倡议(如COVID Moonshot)和数据共享标准(Erlanson et al., 2025关于片段数据最佳实践)是应对这一挑战的策略。
12.4 未来发展方向
闭环自动化实验室(Closed-Loop Autonomous Lab):将机器学习决策、机器人合成和实时实验测试整合为自主迭代的DMT系统。尽管完整实现仍在早期,但多智能体AI实验室自动化框架(Fehlis et al., 2025等arXiv前沿工作)已初显雏形。
端到端数据标准化与共享:建立跨研究机构的片段数据格式标准、注释规范和最佳实践,是训练下一代AI模型、基准测试新方法的基础。Open Reaction Database(Kearnes et al.)和结构数据共享倡议是重要先例。
合成新方法的持续整合:C-H活化、光催化、金属-光氧化还原双催化等方法将持续拓展片段可达的化学空间,与FBDD的结合将更加系统化。
模块化片段与PROTAC/分子胶的协同:片段的模块化特性使其特别适合作为PROTAC中E3配体或底物配体的起始点,以及分子胶筛选的种子,为靶向蛋白降解策略提供新的化学工具。
13. 总结
这篇综述的价值不在于提供某一具体方法的操作细节,而在于以DMT循环为叙事主轴,将FBDD的整个研究生态以动态、整合的视角加以呈现:
1. 系统性框架:厘清了片段从库设计→筛选→命中验证→设计→合成→测试各阶段的逻辑关联,特别强调各阶段之间的信息流动和方法协同 2. 方法论深度:对每一类方法的原理、适用条件、典型局限和前沿进展均有深入讨论,不流于表面 3. 数据驱动案例:通过PDE10A、A1/A3腺苷受体、SARS-CoV-2 Mpro、BRD4等真实案例,将抽象方法框架与具体药物发现成果相连接 4. 批判性视角:对AI/ML的早期局限、粗糙数据的争议、公平获取挑战等问题均坦诚讨论,而非一味唱好
参考文献(精选)
以下精选部分具有代表性的原始引用,完整参考文献共详见原文。
• Grosjean H. et al. Binding-Site Purification of Actives (B-SPA) enables efficient large-scale progression of fragment hits.Angew. Chem. Int. Ed. (2025) • Layton M.E. et al. Discovery of MK-8189, a highly potent and selective PDE10A inhibitor.J. Med. Chem. 66, 1157 (2023) • Boby M.L. et al. Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors.Science 382, eabo7201 (2023) • Yu H.S. et al. General theory of fragment linking in molecular design.J. Chem. Theory Comput. 17, 450 (2021) • Chessari G. et al. C–H functionalisation tolerant to polar groups could transform FBDD.Chem. Sci. 12, 11976 (2021) • Douangamath A. et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease.Nat. Comms. 11, 5047 (2020) • Erlanson D.A. et al. Where and how to house big data on small fragments.Nat. Comms. 16, 4179 (2025) • Sadybekov A.V. & Katritch V. Computational approaches streamlining drug discovery.Nature 616, 673 (2023) • Alibay I. et al. Evaluating the use of absolute binding free energy in fragment optimisation.Commun. Chem. 5, 105 (2022) • Khalak Y. et al. Chemical space exploration with active learning and alchemical free energies.J. Chem. Theory Comput. 18, 6259 (2022)
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢