

Stanford CS224W: Machine Learning with Graphs
代码下载:https://t.zsxq.com/J7Ogi
请索引第34个项目
 |  |
为什么我们需要更好的药理学(Pharmacological)关联预测
现代药理学(Modern pharmacology)依赖于药物间的相互作用。一种药物(drug)可以与数十种蛋白质(proteins)结合,治疗多种疾病,引发意想不到的副作用(side effects),或者与其他药物发生危险的相互作用。所有这些关联都存在于不同的数据库中,但报告的可靠性参差不齐,而且常常被研究界孤立看待。因此,发现现有药物的新用途或预测新药可能产生的风险相互作用,仍然需要从各种不同的信息来源中拼凑出完整的信息。

与此同时,我们看到生物医学数据(biomedical data)空前丰富。我们现在拥有大规模的化合物-蛋白质检测、精心整理的临床疗效标签、不良事件报告系统以及新兴的多组学数据集(multi-omics datasets)。然而,尽管数据如此丰富,大多数计算模型对药理学的处理方式却过于狭隘,它们要么预测药物的靶点,要么预测其临床适应症,要么预测其副作用——很少能同时预测所有这些方面。大多数现有模型将药物、蛋白质或效应简化为固定的向量,隐含地假设它们是可比较的对象,而这种假设在生物学上是站不住脚的。本质上,当前的预测工具要么信息匮乏,要么过于简单化。
图 1. 传统药物相互作用 (DDI) 预测流程示例,该流程仅关注单一关系类型。这些模型通常依赖于药物层面的特征和成对相互作用,而没有纳入更广泛的生物医学背景,例如蛋白质靶点、疾病机制或多关系图结构。
正是这种碎片化促使我们开展了这个项目。与其孤立地对每种关系类型进行建模,不如将它们统一到一个单一的异构生物医学知识图谱中?该图谱包含药物、蛋白质、疾病/效应、基因和不良事件等节点,并通过各种生物化学和临床关系连接起来?
原则上,这样的图表可以涵盖完整的药理学概况:
药物→蛋白质边缘编码分子结合,
药物→效应边缘编码治疗用途,
药物→药物边缘编码禁忌症或相互作用,
其他生物医学实体则提供了生物学背景。
在这种结构下,学习不再仅仅是预测,而是基于多模态、多关系证据进行推理。
这就直接引出了我们的研究问题:
与仅包含药物-靶点-效应图的模型相比,整合多关系生物医学数据是否能提高药理学关联预测模型的性能和可解释性?
为了系统地研究这个问题,我们采用了一种渐进式建模策略。首先,我们构建一个包含药物、蛋白质和效应的聚焦三模态图,以在受控条件下建立可靠的基线。然后,我们逐步提高架构的表达能力和图的复杂度,从经典的知识图谱嵌入,到异构图神经网络,最终过渡到模式丰富且注意力驱动的关系模型。
在所有阶段,我们的动机始终如一:超越单一任务药理学模型,走向统一的系统,从而发现新的治疗机会,识别以前未知的药物-靶点相互作用,并最终通过更丰富的关系推理来支持更安全的药物开发。
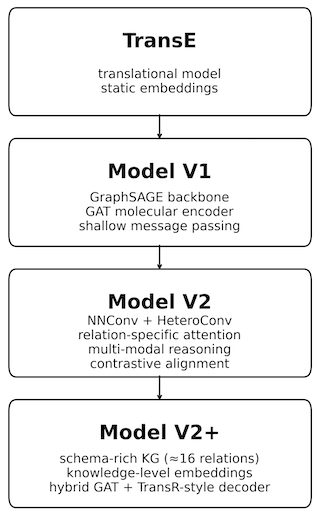
图 2. 药理学联系预测的渐进模型演进,从经典的知识图谱嵌入(TransE)发展到具有关系感知注意力和模式丰富推理的更具表现力的异构图神经网络。
数据集和问题定义
为了探究我们的研究问题,我们以真实的生物医学数据集ChEMBL 36为基础开展工作。ChEMBL是一个大型的、经过精心整理的药物-靶点相互作用数据存储库。虽然我们的长期目标是整合多个生物医学数据源(例如DrugBank、SIDER、FAERS、DisGeNET),但我们的建模策略有意分阶段进行。我们并非固定使用单一的静态图,而是构建一系列药理学图,这些图的模式丰富度和建模复杂度均逐步增加。
我们首先构建一个核心图,在受控条件下建立清晰的基线,然后随着模型表达能力的提升,逐步扩展节点类型、关系类型和表示形式。这种分阶段的设计使我们能够区分图结构、节点表示和关系推理能力对不同模型的影响。
核心三模态药理学图
我们用于 TransE、模型 V1 和模型 V2 的初始图由从 ChEMBL 中提取的三种生物学中心节点类型组成:
3127 个 药物节点,并非以简单的指纹图谱形式表示,而是使用 RDKit 从 SMILES 字符串导出的完整分子图谱。
1156 个蛋白质节点,每个节点均使用来自 ESM-2 蛋白质语言模式的 2560 维嵌入进行编码。
1065 个效应节点,代表治疗或临床结果,使用 32 维可学习嵌入进行建模。
我们提取了原子特征(氢原子数、杂化方式、芳香性、形式电荷、键度和符号)和键特征(键类型),然后应用ESM2编码进行嵌入。每个原子成为一个57维向量,每个键成为一个4维向量。
# Function to compute embeddings in batches
defcompute_esm2_embeddings_batch(sequences, batch_size=4):
"""Compute ESM-2 embeddings for multiple sequences."""
all_embeddings = []
num_batches = (len(sequences) + batch_size - 1) // batch_size
print(f"\nComputing embeddings in {num_batches} batches...")
for i in tqdm(range(0, len(sequences), batch_size)):
batch = sequences[i:i+batch_size]
# Tokenize batch
inputs = tokenizer(
batch,
return_tensors="pt",
truncation=True,
max_length=MAX_LENGTH,
padding=True
)
inputs = {k: v.to(DEVICE) for k, v in inputs.items()}
# Get embeddings
with torch.no_grad():
outputs = model(**inputs)
# Mean pooling over sequence length
embeddings = outputs.last_hidden_state.mean(dim=1) # (batch_size, 1280)
embeddings = embeddings.cpu().numpy()
all_embeddings.append(embeddings)
# Clear GPU memory
del inputs, outputs, embeddings
if DEVICE == "cuda":
torch.cuda.empty_cache()
return np.vstack(all_embeddings)我们将所有药物编码为 PyTorch 几何数据对象,然后对蛋白质也进行了同样的操作。此外,我们还为 1065 个独特的效应创建了独热编码嵌入。在对这三种节点类型进行编码后,我们构建了一个包含药物-蛋白质和药物-效应边的异构图。最终,我们获得了 11703 个药物-蛋白质相互作用和一个完整的图结构,可用于建模。
protein_pyg_objects = []
for idx, row in tqdm(protien_nodes.iterrows(), total=len(protien_nodes), desc="Creating PyG objects"):
embedding_str = row['esm2_embedding']
embedding = np.array(embedding_str)
x = torch.FloatTensor(embedding).unsqueeze(0) # Shape: (1, 1280)
data = Data(
x=x, # node features (just one node per protein)
protein_internal_id=int(row['protein_internal_id']),
protein_id=str(row['protein_id']),
protein_name=str(row['protein_name']),
uniprot_id=str(row['uniprot_id']),
sequence_length=int(row['sequence_length'])
)
protein_pyg_objects.append(data)在这些节点上,我们定义了两种主要的药理学关系类型:
药物 → 蛋白质(结合到)
药物→作用(治疗)
我们使用 PyTorch Geometric 作为主要框架来构建完整的异构药理学模型。第一个组件是使用图注意力网络 (GAT) 的药物分子图编码器。接下来,我们构建了异构图卷积层来处理不同节点类型之间的消息传递。最后,我们组装了整个模型架构,其中包括药物分子编码器、蛋白质和效应投影、多个异构图卷积层以及用于药物-蛋白质和药物-效应预测的链接预测头。
classPharmacologyHeteroGNN(nn.Module):
"""
Complete heterogeneous GNN for pharmacology knowledge graph
Architecture:
1. Drug molecular encoder (GAT) → shared_dim
2. Protein projection (ESM2 embeddings) → shared_dim
3. Effect projection (learnable embeddings) → shared_dim
4. Multiple heterogeneous graph conv layers
5. Link prediction heads for drug-protein and drug-effect
"""
definit(self, config):
super(PharmacologyHeteroGNN, self).init()
self.config = config
shared_dim = config['shared_dim']
# 1. Molecular graph encoder for drugs
self.drug_molecular_encoder = DrugMolecularEncoder(
node_feat_dim=config['drug_node_feat_dim'],
edge_feat_dim=config['drug_edge_feat_dim'],
hidden_dim=config['drug_hidden_dim'],
output_dim=shared_dim,
num_layers=config['drug_num_layers'],
heads=config['drug_heads']
)
# 2. Projections for protein and effect features
self.protein_proj = nn.Sequential(
nn.Linear(config['protein_feat_dim'], shared_dim),
nn.LayerNorm(shared_dim),
nn.ReLU(),
nn.Dropout(0.1)
)
self.effect_proj = nn.Sequential(
nn.Linear(config['effect_feat_dim'], shared_dim),
nn.LayerNorm(shared_dim),
nn.ReLU(),
nn.Dropout(0.1)
)
# 3. Heterogeneous graph convolution layers
edge_types = [
('drug', 'binds_to', 'protein'),
('protein', 'rev_binds_to', 'drug'),
('drug', 'treats', 'effect'),
('effect', 'rev_treats', 'drug')
]
self.hetero_layers = nn.ModuleList([
HeteroGraphConvLayer(shared_dim, edge_types)
for _ inrange(config['num_hetero_layers'])
])
# 4. Link prediction heads
self.drug_protein_predictor = nn.Sequential(
nn.Linear(shared_dim * 2, shared_dim),
nn.ReLU(),
nn.Dropout(0.2),
nn.Linear(shared_dim, 1)
)
self.drug_effect_predictor = nn.Sequential(
nn.Linear(shared_dim * 2, shared_dim),
nn.ReLU(),
nn.Dropout(0.2),
nn.Linear(shared_dim, 1)
)由此生成了一个包含超过18,000 条带标签边的异构图,涵盖了分子结合和治疗背景。即使在现阶段,该图也超越了单一关系管道,将多个生物学轴、化学结构、蛋白质生物学和临床结果嵌入到一个统一的关系框架中。
扩展的模式丰富建模图(V2+)
在最终的探索性模型(V2+)中,我们将重点从增加表示深度转移到扩展图模式的广度。在此设置下,我们通过引入额外的节点和关系类型来大幅扩展图,包括:
作用机制靶点
药物警告
分层治疗分类节点(多级)
其他生物医学关系(总共约 16 种关系类型)
from torch_geometric.data import Data, HeteroData
# Each drug is its own molecular graph (atoms attend only to atoms in same molecule)
drug_pyg_objects = []
for drug in drug_graph_embeddings:
data = Data(
x=torch.FloatTensor(drug['node_attr']), # (num_atoms, 65)
edge_index=torch.LongTensor(drug['edge_index']),
edge_attr=torch.FloatTensor(drug['edge_attr']),
drug_internal_id=drug['drug_internal_id']
)
drug_pyg_objects.append(data)
# Knowledge graph edges stored as edge_index tensors per relation type
full_edge_index_dict = {
('drug', 'binds_protein', 'protein'): drug_protein_edge_index,
('protein', 'bound_by_drug', 'drug'): drug_protein_edge_index.flip(0),
('drug', 'causes_effect', 'effect'): drug_effect_edge_index,
('effect', 'caused_by_drug', 'drug'): drug_effect_edge_index.flip(0),
# ... 16 total edge types (see next section)
}重要的是,这个扩展图尚未包含明确的药物相互作用(DDI)边。由于内存和基础设施的限制,V2+ 基于知识层嵌入而非分子图编码器运行,这使我们能够研究仅通过模式扩展就能在不进行深度化学或序列建模的情况下,将关系推理能力提升到何种程度。
问题定义:多关系药理学链接预测
我们将任务定义为异构链接预测:
药物→蛋白质——该模型能否推断出先前未知的分子结合关系?
药物→效应——能否发现潜在的治疗适应症或重新利用的候选药物?
(未来扩展) 药物 → 药物—能否预测不良相互作用或禁忌症?
这些任务都受益于共享的图结构。通过在药物、蛋白质和效应节点之间传播信息,该模型可以利用孤立方法所忽略的通路。例如,如果某种药物与已与该效应相关的蛋白质结合,则可以推断该药物可能治疗某种疾病。
我们在转导式设置下评估了我们的模型,这意味着所有节点在训练过程中都是可见的,但特定的边则保留用于测试。这种设置反映了实际的药理学问题:在掌握所有现有的生物医学知识的情况下,我们能否预测尚未发现或实验验证的缺失关系?
我们的模型演进:从简单的嵌入到深度关系推理
为了了解准确药理预测需要多强的模型表达能力,我们采用了一种迭代建模策略。我们没有预先提出单一的架构,而是在保持核心药理学目标不变的情况下,逐步探索设计空间中的不同点,改变表征深度和关系表达能力。
该序列中的每个模型都旨在回答一个特定问题,即在对生物医学图进行推理时,什么才是最重要的。
1. TransE — 经典知识图谱嵌入基线
我们首先介绍TransE,这是一种广泛应用于知识图谱研究的翻译嵌入模型。在该模型中,药物、蛋白质和效应被表示为静态嵌入,而关系则被建模为潜在空间中的翻译。
尽管TransE算法简单,但它在我们精心构建的密集核心图谱上表现出了令人惊讶的强大区分能力。这一结果凸显了精心构建的药理学关系中已蕴含着大量信号。然而,TransE也存在一些根本性的局限性,这些局限性在生物医学领域尤为突出:
它难以处理多对多关系(在药物-靶点相互作用中很常见),
它依赖于静态嵌入,无法适应不同的关系类型。
它无法整合分子结构或蛋白质序列等多模态特征。
因此,TransE 可以作为一个方向点:它确定了纯粹的关系结构在需要更丰富的生物学表征之前能带我们走多远。
2. 模型 V1 — GraphSAGE 基线模型,采用 GAT 分子编码器
我们的第二个模型通过异构图神经网络引入了具有生物学意义的节点表示。在模型 V1 中,我们整合了:
用于处理药物分子图的GAT编码器,
冷冻ESM-2蛋白包埋物,
以及一个采用均值聚合的GraphSAGE 消息传递骨干网,然后是用于链路预测的 MLP 解码器。
该模型首次引入了归纳偏置和结构感知,使得信息能够在药物、蛋白质和效应节点之间传播。然而,性能提升趋于平缓,尤其是在药物-效应预测方面。经验表明,这揭示了一个重要的局限性:尽管GraphSAGE具有良好的可扩展性,但当存在多种生物医学关系类型时,其平均聚合会削弱关系特异性。
模型V1表明,仅添加节点级生物学特征是不够的。有效的药理学推理还需要对关系类型进行更具表现力的处理。
3. 模型 V2 — 注意力增强型神经网络卷积架构
模型 V2 是我们的主要贡献,它通过大幅提升关系表达能力,解决了早期模型存在的局限性。该架构结合了以下几点:
用于边缘条件分子信息传递的NNConv ,
基于注意力机制的异构卷积可以显式地对不同类型的关系进行加权,
对比学习将药物、蛋白质和效应嵌入对齐到共享的潜在空间中。
在这个更新后的模型中,我们不再将边视为静态链接,而是让它们主动塑造消息在网络中的流动方式。每条边的特征有助于调节节点间传递的信息,从而实现更丰富、更具上下文感知的编码。通过对这一过程进行分层处理,并应用诸如dropout之类的正则化技术,我们旨在确保这些嵌入既稳健又富有表现力。
classDrugMolecularEncoder(nn.Module):
"""Encode drug molecular graphs using NNConv for edge-conditioned message passing"""
definit(self, node_feat_dim, edge_feat_dim, hidden_dim, output_dim, num_layers=3, heads=4):
super(DrugMolecularEncoder, self).init()
self.node_encoder = nn.Linear(node_feat_dim, hidden_dim)
# Edge network for NNConv - transforms edge features into message weights
self.edge_networks = nn.ModuleList()
self.nn_convs = nn.ModuleList()
for i inrange(num_layers):
in_dim = hidden_dim
# Small MLP that takes edge features and outputs weight matrix for message passing
edge_net = nn.Sequential(
nn.Linear(edge_feat_dim, hidden_dim * hidden_dim),
nn.ReLU(),
nn.Linear(hidden_dim * hidden_dim, in_dim * hidden_dim)
)
self.edge_networks.append(edge_net)
self.nn_convs.append(NNConv(in_dim, hidden_dim, edge_net, aggr='mean'))
self.output_proj = nn.Linear(hidden_dim, output_dim)
self.dropout = nn.Dropout(0.1)接下来,我们引入注意力机制来处理图的异构性。由于我们处理两种类型的边,因此我们使用多头注意力机制,让模型学习在给定上下文中哪些关系最为重要。
classAttentionHeteroConv(nn.Module):
"""Attention-based heterogeneous convolution with interpretable aggregation"""
definit(self, hidden_dim, edge_types, num_heads=4):
super(AttentionHeteroConv, self).init()
self.hidden_dim = hidden_dim
self.num_heads = num_heads
self.head_dim = hidden_dim // num_heads
# Message passing layers for each edge type
self.convs = nn.ModuleDict()
for edge_type in edge_types:
src_type, rel, dst_type = edge_type
key = f"{src_type}{rel}{dst_type}"
self.convs[key] = SAGEConv((hidden_dim, hidden_dim), hidden_dim, aggr='mean')
# Multi-head attention for aggregating different edge types
self.query_proj = nn.Linear(hidden_dim, hidden_dim)
self.key_proj = nn.Linear(hidden_dim, hidden_dim)
self.value_proj = nn.Linear(hidden_dim, hidden_dim)
self.out_proj = nn.Linear(hidden_dim, hidden_dim)通过使用 InfoNCE 损失函数,我们鼓励来自不同节点类型(例如药物和蛋白质或药物和效应)的嵌入向量在应该相似的情况下更加紧密地对齐。我们试图从根本上加强相关节点之间的对齐,并提高链接预测的整体质量。
defcontrastive_loss(embeddings_dict, temperature=0.5):
"""InfoNCE contrastive loss to align embeddings across node types"""
# Get embeddings from different node types
drug_emb = embeddings_dict.get('drug', None)
protein_emb = embeddings_dict.get('protein', None)
effect_emb = embeddings_dict.get('effect', None)
if drug_emb isNoneor (protein_emb isNoneand effect_emb isNone):
return torch.tensor(0.0, device=drug_emb.device if drug_emb isnotNoneelse'cpu')
loss = 0.0
count = 0
# Contrastive loss between drug and protein embeddings
if protein_emb isnotNoneand drug_emb.size(0) > 1:
# Normalize embeddings
drug_norm = F.normalize(drug_emb, p=2, dim=1)
protein_norm = F.normalize(protein_emb, p=2, dim=1)
# Compute similarity matrix
logits = torch.mm(drug_norm, protein_norm.t()) / temperature
# Create positive pairs (assume diagonal are positives)
min_size = min(drug_norm.size(0), protein_norm.size(0))
labels = torch.arange(min_size, device=drug_emb.device)
# Cross entropy loss (InfoNCE)
loss_dp = F.cross_entropy(logits[:min_size], labels)
loss += loss_dp
count += 1
# Contrastive loss between drug and effect embeddings
if effect_emb isnotNoneand drug_emb.size(0) > 1:
drug_norm = F.normalize(drug_emb, p=2, dim=1)
effect_norm = F.normalize(effect_emb, p=2, dim=1)
logits = torch.mm(drug_norm, effect_norm.t()) / temperature
min_size = min(drug_norm.size(0), effect_norm.size(0))
labels = torch.arange(min_size, device=drug_emb.device)
loss_de = F.cross_entropy(logits[:min_size], labels)
loss += loss_de
count += 1
return loss / max(count, 1)与之前的模型不同,模型 V2 允许网络根据每种关系的生物学语义来区分信息的传播方式。这使得模型性能相比 TransE 和模型 V1 有了显著提升,尤其是在药物-蛋白质预测方面,并且整体精度和排序质量也得到了显著提高。
这些成果表明,药理学从根本上来说是多关系的,而具有表达能力的、感知关系的架构对于捕捉连接分子结构、蛋白质功能和临床结果的多跳生物依赖性至关重要。
4. 新兴方向——V2+ 和富含模式的药理学图谱
模型 V2 侧重于增加表示深度,而我们最终的探索性模型(V2+)则研究了一个互补的问题:扩展图模式本身能给我们带来多少好处?
在 V2+ 版本中,我们通过引入额外的节点和关系类型(总共约 16 种)大幅扩展了图的广度,其中包括:
作用机制靶点,
药物警告
以及分层治疗分类节点。
由于内存和基础设施的限制,V2+ 基于知识级嵌入而非分子图编码器运行,并且尚未包含显式的药物相互作用边。在架构上,它采用了一种混合的 GAT + TransR 式设计,无需完整的全局注意力机制或位置编码即可实现特定关系的投影。
V2+ 并非取代模型 V2,而是探索设计空间的另一个维度:模式丰富度与表征深度。该模型有助于分离更广泛的生物医学背景的影响,使其独立于深度化学或序列建模,并为未来完全基于注意力机制的药理学图模型奠定基础。
模型结果与见解
我们建模流程的每个阶段都揭示了有效药理学推理所需要素的不同方面。我们的结果并非在单一静态图上评估所有模型,而是体现了一种渐进式的实验设计:TransE、模型 V1 和模型 V2 在同一个核心三模态图上进行评估,而模型 V2+ 则在一个扩展的模式图上进行评估,该模式图旨在探究关系广度。
在这些场景下,我们主要使用AUC-ROC评估模型,并在适当情况下辅以精确率、召回率、F1值和排名指标。AUC尤其适用于生物医学关联预测,因为其中负样本的数量远远超过正样本,并且AUC能够反映模型在不同阈值下区分真实关联和错误关联的能力。
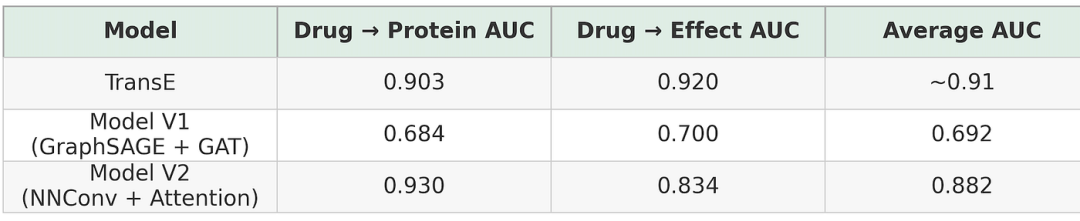
图 3.三种建模方法的性能比较:TransE、基于 GraphSAGE 的异构 GNN(模型 V1)和注意力增强的 NNConv 模型(模型 V2)。
1. TransE 简单、强大且极具竞争力
尽管 TransE 最初并非为生物医学图谱而设计,但它在我们的数据集上却展现出了令人惊讶的强大基线能力。其正负分值分布清晰分离,AUC 值(≈0.90+)表明其具有很强的区分能力。
然而,这种表面上的优势掩盖了其重要的局限性:
尽管AUC值较高,但排名质量较差(MRR ≈ 0.29)。
药理学中普遍存在的多对多关系建模存在困难。
无法适应不同关系类型或包含生物结构的静态实体嵌入
图 4. TransE 模型学习到的节点嵌入的二维 t-SNE 投影。虽然正负关系大致可以分离,但嵌入空间呈现出粗糙的聚类和有限的局部结构,这反映出 TransE 使用的是静态实体嵌入,缺乏关系感知或多模态表达能力。
如图4所示,TransE 能够捕捉粗略的关系信号,但无法将药物组织成精细的、具有机制意义的邻域。这些结果表明,虽然精心构建的药理学图谱包含大量表面信号,但纯粹的转化嵌入不足以进行更深层次的生物学推理。
2. 模型V1揭示了浅层消息传递的局限性
我们的第一个 GNN(模型 V1)通过 GAT 编码器和 GraphSAGE 骨干网络引入了归纳学习和分子结构感知。它首次整合了多模态特征,但在某些指标上性能仅略有提升,整体性能不如 TransE。
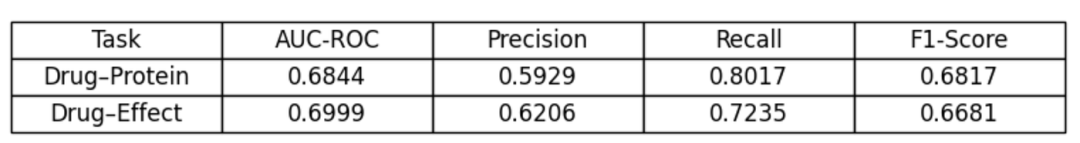
图 5.模型 V1 统计结果
为什么?
GraphSAGE的平均聚合稀释了关系特异性。来自不同邻居类型的信号混合在一起,使得模型难以区分生化通路和临床通路。
GAT 在分子层面有所帮助,但下游信息传递的表达能力不足以推断多种生物医学实体类型之间的相互作用。
效果预测仍然特别困难,这表明临床结果需要比 V1 能够提取的更深层次的背景信息。
模型 V1 告诉我们,并非所有图神经网络 (GNN) 都足以表达异构生物医学图,尤其是那些关系归纳偏置较浅的图神经网络。它表明,添加具有生物学意义的节点特征是必要的,但对于有效的药理学推理而言并非充分条件。
3. V2 型号的表现力改变了一切
模型 V2 通过大幅提升关系表达能力,克服了早期模型的局限性。它结合了以下特点:
用于边缘条件分子信息传递的NNConv
基于注意力机制的异构卷积用于关系特定聚合
对比学习以统一药物、蛋白质和效应表征
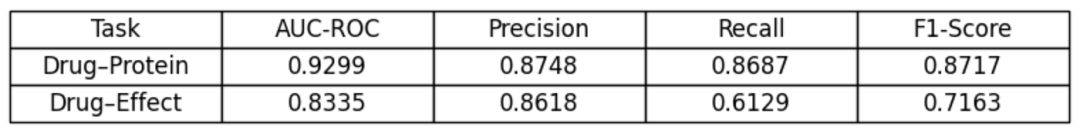
图 6.模型 V2 统计结果
这些改变带来了显著的性能提升:
药物-蛋白质预测AUC增加至0.93
与模型 V1 相比,总体平均 AUC 提高了近20 个百分点。
精确率、召回率和 F1 分数持续提高
为什么这很重要?因为药理学本质上是多层次的关联过程。药物的治疗效果不仅取决于分子结合,还取决于下游生物通路及其与临床结果的关系。模型 V2 是我们流程中第一个能够充分表达这种多级推理的架构。
尤其:
注意力机制使模型能够将binds_to和treats视为生物学上不同的关系。
NNConv在分子水平上保留了化学性质的忠实表示。
对比对齐有助于在分子、生物和临床嵌入空间之间保持一致性。
这些组成部分共同产生的表征反映了药理学的相互关联性,而不是将其简化为一个单一的潜在空间。
4. 模型 V2+ — 基于 TransR 知识嵌入的模式扩展
虽然模型 V2 展示了表征深度和关系感知消息传递的重要性,但我们的最终模型(V2+)探索了一个不同的问题:即使没有深度分子或序列编码器,我们也能从扩展图模式本身中获得多少收益?
为了分离模式丰富度的影响,我们采用了一种TransR风格的知识图谱嵌入模型,该模型运行在一个大幅扩展的药理学图谱上。该图谱包含了诸如acts_on_target、has_warning和has_therapeutic_class等额外的关系类型,总共产生了大约16种关系类型。
由于GPU内存的限制,我们特意将此模型限制为仅使用实体和关系嵌入,而非分子图编码器或全局Transformer注意力机制。这种设计选择使我们能够将图扩展到近100万个三元组和超过190万个实体,同时清晰地隔离关系广度的影响。
尽管架构简化,V2+ 仍实现了强大的性能:
AUC-ROC: 0.894
F1 分数: 0.813
平均排名: 8.1
点击量@10: 0.752
这些结果表明,即使没有深度神经网络编码器,仅模式扩展也能提供显著的预测信号。与之前的 TransE 基线相比,TransR 的关系特定投影空间显著提高了排序质量和校准度。
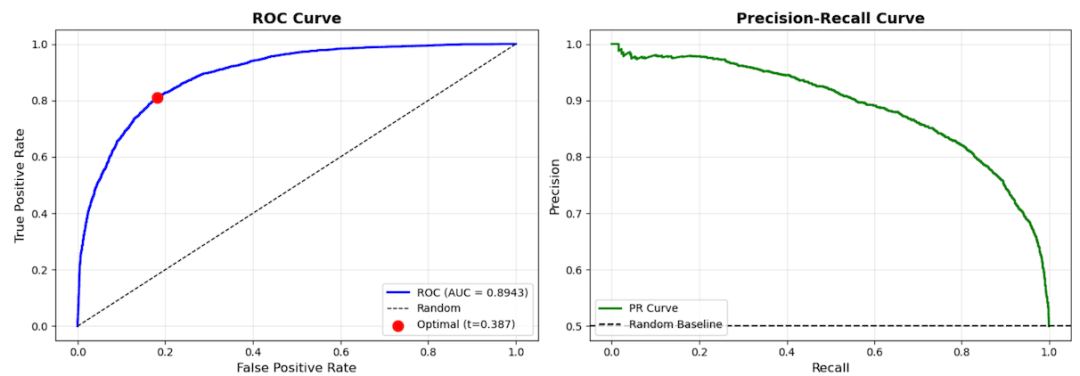
图 7. V2+ 模型的 ROC 曲线和精确率-召回率曲线
扩展模式的一个关键优势是能够评估每个关系的行为,从而揭示模式的丰富性在哪些方面有所帮助,在哪些方面没有帮助。
高效关系
药物与蛋白质结合:AUC ≈ 0.90
药物作用效应:AUC ≈ 0.84
药物作用于靶点:AUC ≈ 0.76
充满挑战的关系
drug_has_warning:AUC ≈ 0.46
drug_has_therapeutic_class : AUC ≈ 0.65
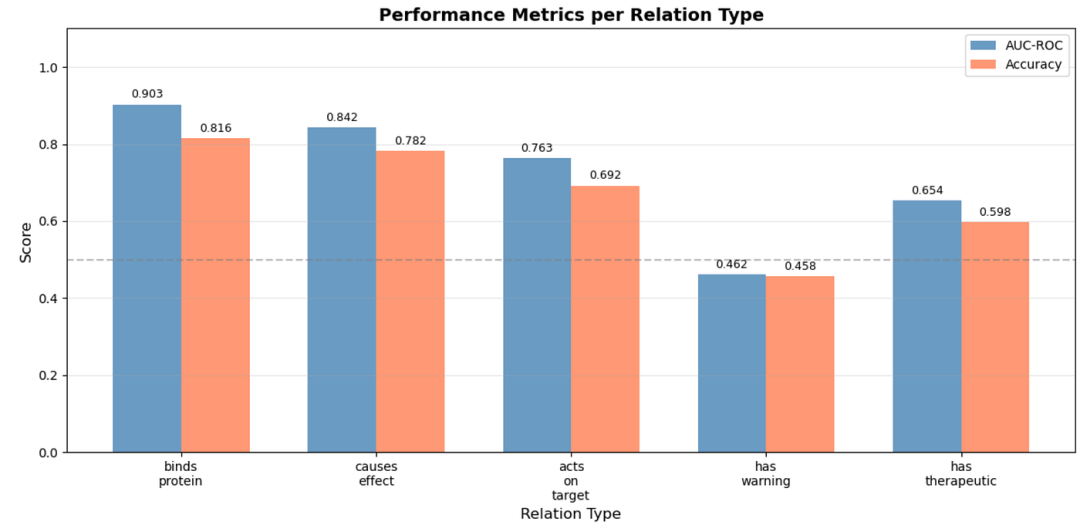
图 8.按关系类型细分 AUC 和准确率
这些结果既展现了生物体的复杂性,也体现了数据的稀疏性。警示标签和治疗分类粗略、嘈杂,且通常与分子机制的关联性较弱,因此仅凭关系结构很难对其进行预测。
为了更好地理解 TransR 如何在扩展图中组织实体,我们使用 t-SNE 可视化学习到的嵌入。即使没有显式的模态编码器,药物、蛋白质和效应也形成了部分可分离的聚类,这表明关系特定的投影空间有助于保留语义结构。
然而,与模型 V2 相比,这些聚类的组织性较差,这强化了一个重要的结论,即模式扩展可以改善覆盖范围和上下文,但不能完全取代表达能力强、感知模态的编码器。
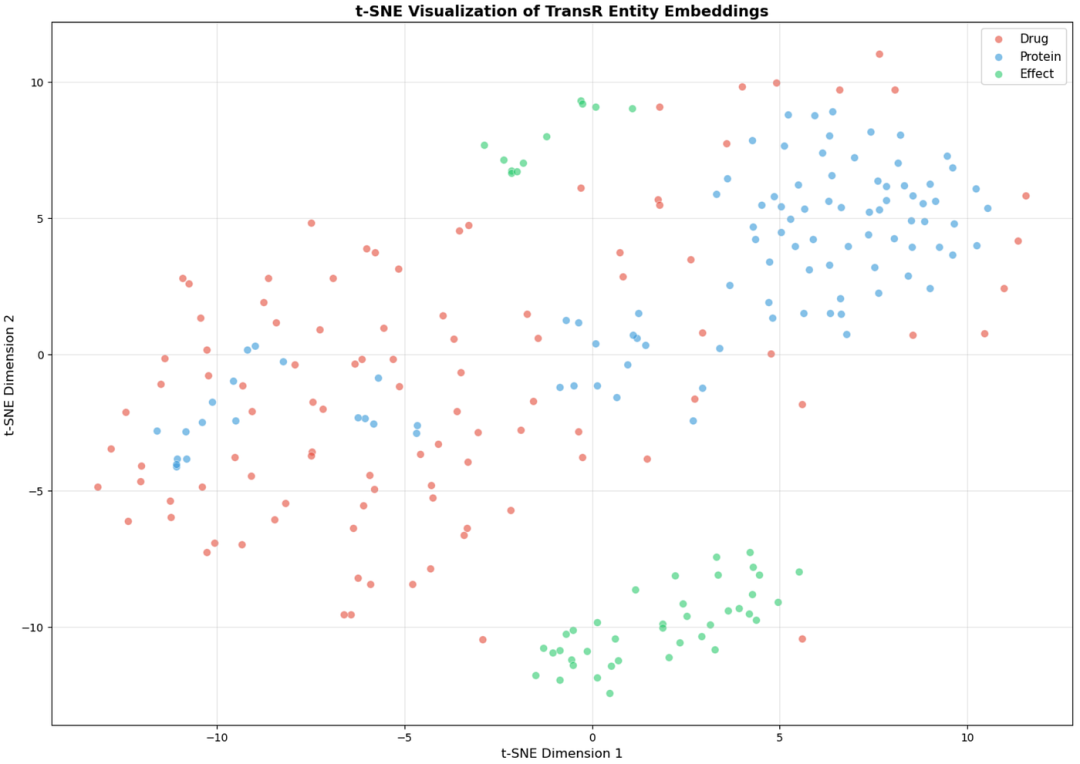
图 9. TransR 实体嵌入的 t-SNE 可视化
模型 V2+ 揭示了药理图建模中的一个关键权衡:
模式广度可以提高关系覆盖率和排序质量,但深入的生物学推理仍然需要具有表达能力、能够感知模态的架构。
这一洞察促使我们自然而然地迈出了下一步,即在内存限制允许的情况下,将模式丰富的图与基于注意力或Transformer风格的模型结合起来。
药物-蛋白质相互作用预测比药物-效应预测更容易
在所有模型中,都出现了一个一致的模式:药物→蛋白质预测更容易。
从生物学角度来看,这是合理的:
蛋白质结合试验更加标准化,噪声更小。
药物-效应关系(治疗适应症)在临床上很复杂,通常是多因素的,而且很少是与结合事件直接相关的。
效应反映的是系统层面的行为,而不仅仅是分子亲和力。
这一见解对于未来的建模至关重要:药物效应预测可以从更丰富的多模态数据和更深层次的关系推理中获得更多收益,而这正是基于 Transformer 的架构的优势所在。
统一图才是真正的超级大国
综合所有三个模型,最有力的结论是:整合多关系生物医学数据使模型更智能。
即使是最简单的模型,一旦我们跳出单一任务框架的局限,也能从中受益。随着多模态特征和架构表达能力的扩展,性能显著提升。
这直接回答了我们的研究问题:
是的,与仅包含药物-靶点-效应或单任务图的图相比,整合多关系生物医学数据可以提高预测性能和可解释性。
参考文献
A. Bordes, N. Usunier, A. Garcia-Duran, J. Weston, and O. Yakhnenko. Translating embeddings for modeling multi-relational data. In Advances in Neural Information Processing Systems, 2013.
M. Schlichtkrull, T. Kipf, P. Bloem, R. van den Berg, I. Titov, and M. Welling. Modeling relational data with graph convolutional networks. In European Semantic Web Conference, 2018.
S. Vashishth, S. Sanyal, V. Nitin, and P. Talukdar. Composition-based multi-relational graph convolutional networks. In International Conference on Learning Representations, 2020.
ChEMBL Team. ChEMBL 36 release notes and blog, 2025. https://chembl.blogspot.com/2025/09/chembl-36-is-out.html.
D. S. Wishart et al. DrugBank 5.0: a major update. Nucleic Acids Research, 46(D1), 2018.
J. Kuhn et al. The SIDER database of drugs and side effects. Nucleic Acids Research, 44(D1), 2016.
FDA Adverse Event Reporting System (FAERS). 2024.
LINCS Consortium. The L1000 data set. Cell Systems, 2017.
J. Pinero et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Research, 48(D1), 2020.
H. Luo et al. Drug-drug interactions prediction based on deep learning and knowledge graph: a review. iScience, 27(3), 109148, 2024.



内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢