
DRUGONE
蛋白质语言模型(protein language models, pLMs)正在迅速改变蛋白质研究领域。从蛋白结构预测、蛋白功能注释,到酶设计与药物发现,越来越多的任务开始依赖深度学习模型完成。然而,这些模型往往像“黑箱”一样运行,研究人员虽然能够获得高精度预测,却难以理解模型究竟依据什么做出判断。模型内部究竟学习了哪些生物学规律、哪些模式真正参与了预测,以及这些规律是否具有真实的生物学意义,仍然是当前人工智能蛋白研究中的核心问题。
为了解决这一问题,可解释人工智能(Explainable Artificial Intelligence, XAI)开始成为蛋白人工智能领域的重要方向。XAI试图通过分析模型内部结构、输入特征以及训练数据,揭示深度学习模型的决策依据,从而提升模型的透明性、可信度与可控性。文章系统梳理了近年来XAI在蛋白质语言模型中的应用,并按照建模流程中的四类信息来源进行分类,包括训练数据、输入序列、模型内部组件以及输入—输出关系。研究人员进一步总结了XAI在蛋白研究中的五种潜在角色:Evaluator(评估者)、Multitasker(多任务者)、Engineer(工程师)、Coach(教练)以及Teacher(导师)。其中,目前最广泛使用的仍然是“评估者”角色,即验证模型是否学习到了研究人员预期中的生物学规律。
文章认为,未来真正具有革命性意义的方向,是让XAI从“验证模型”走向“发现新生物学规律”,即实现Teacher角色。这意味着AI不仅能够预测蛋白性质,还能够帮助研究人员理解蛋白折叠、酶催化以及蛋白进化背后的深层机制。

蛋白语言模型为何需要“可解释性”
随着DNA测序成本下降以及蛋白结构数据库的迅速扩张,蛋白质语言模型的发展进入了高速阶段。早期的pLM主要基于蛋白序列训练,而最新模型则开始融合结构信息、功能注释以及多模态生物学数据。类似于自然语言处理中对文本进行tokenization,蛋白模型也需要将氨基酸序列、结构以及功能信息编码为适合神经网络处理的数学表示。
然而,训练数据本身存在大量偏差。例如,不同物种之间的数据分布不均衡、某些蛋白家族被过度采样、实验技术差异导致的数据偏倚等,都可能影响模型学习结果。此外,许多蛋白模型实际上仍然高度依赖进化同源信息。虽然pLM最初被设计为不依赖MSA(multiple sequence alignment)的单序列模型,但研究表明,在许多任务中,模型性能仍然与MSA深度高度相关。
与此同时,模型规模不断扩大也使得其内部机制越来越难以解释。尽管AlphaFold、ESMFold等模型能够取得惊人的预测精度,但研究人员往往无法回答一个关键问题:模型究竟“理解”了什么?它是真正学习到了物理化学规律,还是仅仅依赖统计相关性完成预测?这一问题在化学语言模型中已经被观察到,即模型可能只是记忆训练数据中的统计规律,而并未真正掌握化学规则。文章认为,蛋白语言模型可能同样面临这一问题。
因此,XAI的重要性不仅在于提高模型可解释性,更在于帮助研究人员判断模型是否真正学习到了具有生物学意义的规律。特别是在蛋白设计、酶工程以及生物安全相关任务中,理解模型内部机制对于提升可靠性与安全性至关重要。
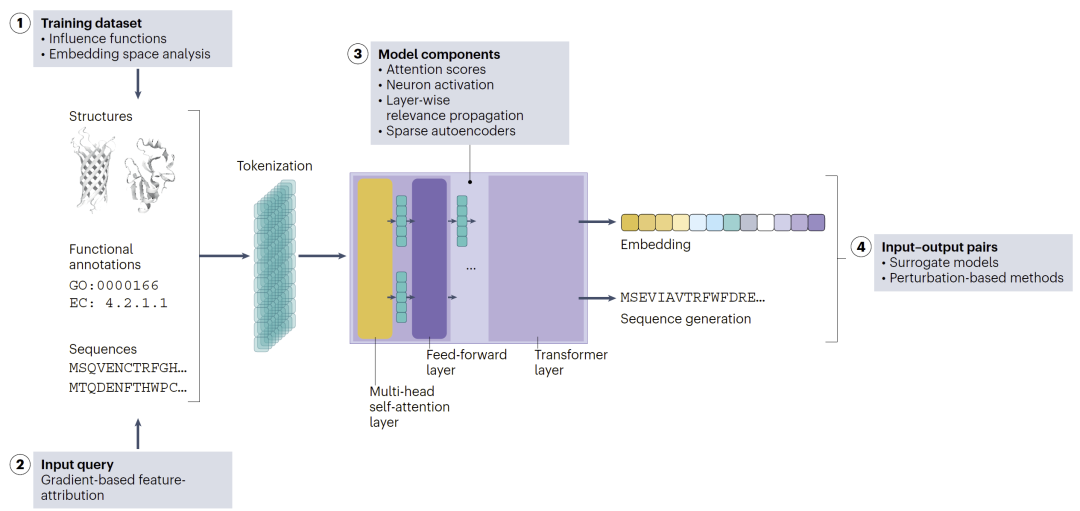
图1:XAI在蛋白质建模流程中的总体框架。
训练数据中的可解释性
研究人员首先讨论了基于训练数据的可解释性分析方法。其中最重要的方法之一是Influence Functions(影响函数)。该方法通过分析训练集中某条序列被移除或权重被改变后,对模型预测结果产生的影响,从而识别哪些训练样本真正决定了模型行为。
研究发现,在蛋白语言模型中,最具影响力的训练数据通常是与目标蛋白具有高度同源性的序列,而且这些影响样本往往服从幂律分布。这意味着只有少数训练样本真正主导了模型的泛化能力。研究人员还发现,对于野生型序列概率过高或过低的情况,模型性能都会下降,因此在低概率区域进行无监督微调反而更有效。
另一种重要方法是Embedding Space Analysis(嵌入空间分析)。研究人员将训练序列和生成序列投影到潜在空间中,通过测量embedding距离分析序列之间的关系。这种方法能够帮助研究人员理解模型如何组织蛋白功能空间,并发现潜在的功能关联。
例如,SeqVec embedding曾被用于基于邻近关系预测Gene Ontology功能注释,而NLP领域中的exBERT则进一步提供了可视化界面,可以同时观察embedding邻近关系与attention map,从而实现多尺度解释。文章认为,未来这些方法能够与数据泄漏分析、训练集偏差分析结合,帮助研究人员更加系统地理解模型行为。
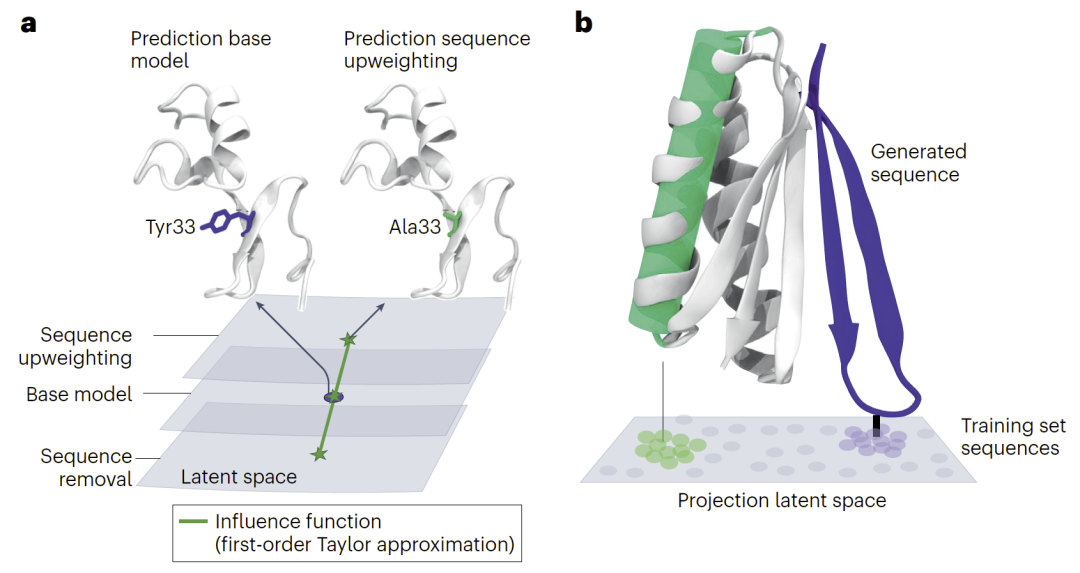
图2:Influence Function与Embedding空间分析示意图。
输入序列中的可解释性
针对输入序列的解释方法主要依赖梯度归因(gradient-based feature attribution)。该类方法通过计算输入特征对模型输出的梯度贡献,量化每个氨基酸残基的重要性。
这些方法已经被用于蛋白无序区预测、抗体亲和力预测等任务。研究人员将每个残基的归因分数映射回原始序列,并与已知生物物理规律进行比较,例如疏水性或结合位点。结果发现,模型确实能够自动识别关键功能残基,并区分重要与非重要位置。
文章进一步提出,在生成模型中,下一氨基酸生成本质上也是一种预测任务,因此可以通过归因方法分析哪些残基决定了后续生成结果。沿整个序列展开后,甚至可能揭示共进化网络或变构调控网络。特别是对于融合结构与功能信息的多模态模型,XAI有望直接揭示残基、结构与功能之间的复杂关联。
此外,研究人员还提出一种更进一步的思路:将生物学先验直接融入loss function。例如,在训练过程中奖励模型关注催化位点,同时惩罚模型学习无意义偏差。这种思路已经在计算机视觉领域取得成功,未来可能迁移到蛋白语言模型中。
不过,梯度归因方法也存在局限。不同方法之间往往会给出不一致的结果,而transformer结构的复杂性也使得传统神经网络解释方法难以直接适用。此外,蛋白语言模型中的离散token与连续embedding之间也存在解释困难,因此目前许多方法实际上是在embedding空间中完成解释,而非直接在氨基酸层面。
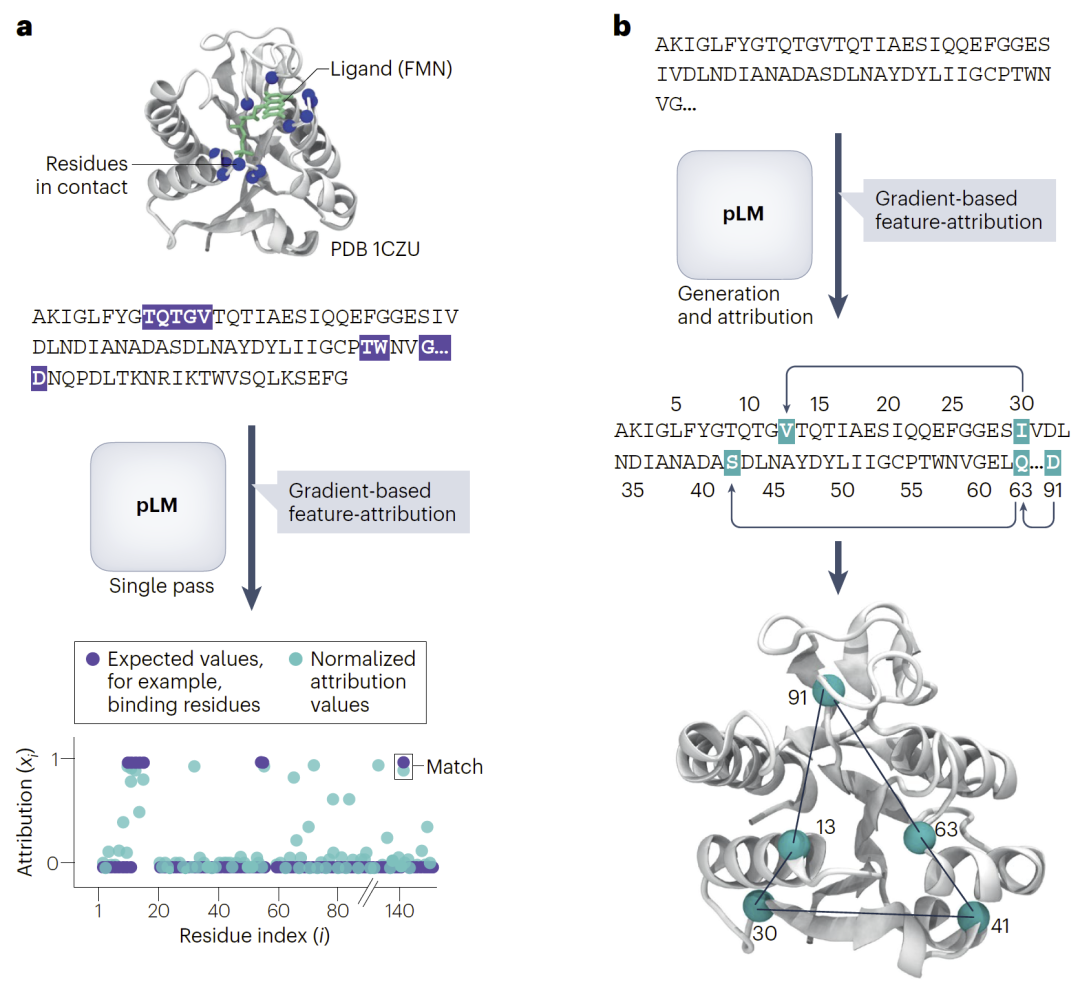
图3:基于梯度归因的残基重要性分析。
模型内部组件的可解释性
由于蛋白语言模型大多采用transformer架构,因此attention机制成为最常见的解释对象。研究人员通常分析attention score是否与真实生物学规律一致,例如结合位点、接触图、氢键或变构位点等。
例如,有研究发现,某些attention head能够专门捕获蛋白接触图,而另一些head则对翻译后修饰高度敏感。这种现象类似于NLP中的“specialized heads”,即某些attention head专门负责语法、位置或特殊语义。
然而,文章也指出,仅仅观察attention并不意味着模型真正依赖attention完成预测。许多研究发现,即使删除部分attention head,模型性能也几乎不受影响。这意味着attention可能只是“相关”,而并非真正“因果”。
因此,研究人员开始探索更深层的解释方法,例如Layer-wise Relevance Propagation(LRP)以及Sparse Autoencoder(SAE)。LRP通过反向传播相关性分数,分析每个神经元对最终输出的贡献。而SAE则试图将高度混合的polysemantic neuron拆分为具有明确意义的monosemantic feature,从而提高可解释性。
近年来,SAE已经开始应用于蛋白语言模型。研究发现,某些latent feature与蛋白家族、酶活性甚至残基疏水性高度相关。更重要的是,这些feature不仅能够解释模型,还能够用于“控制”模型。例如,通过增强与疏水性相关的feature,可以引导ESMFold生成不同结构;通过增强与酶活性相关的feature,则能够生成更高活性的alpha-amylase。
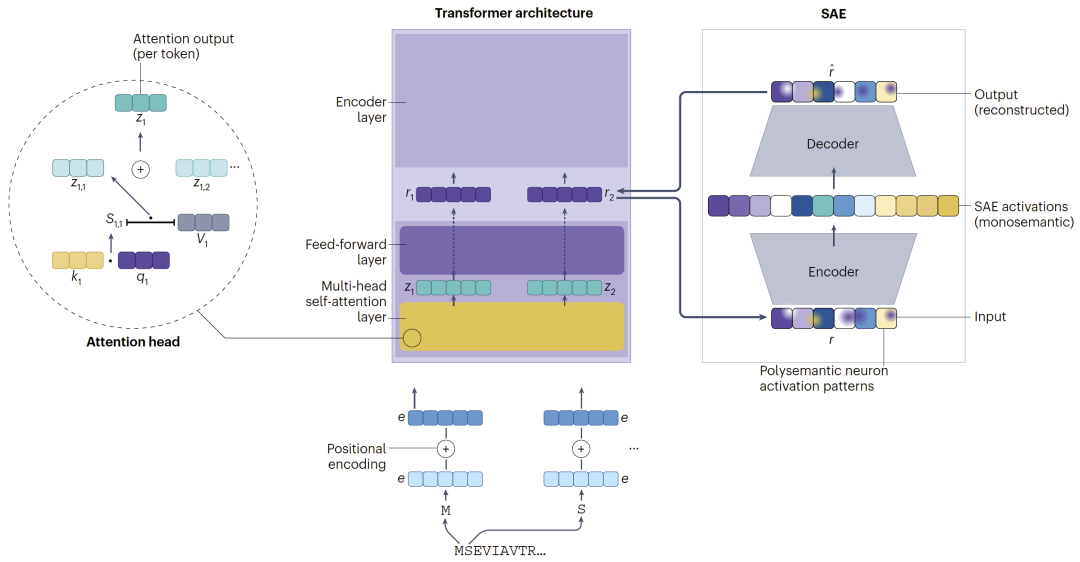
图4:Transformer与Sparse Autoencoder解释框架。
输入—输出关系中的可解释性
另一类重要方法是基于输入—输出关系的扰动分析。研究人员通过系统性修改输入序列,并观察输出变化,从而推断模型的决策逻辑。
其中最常见的方法包括SHAP与LIME。SHAP通过采样不同特征组合估计每个残基的重要性,而LIME则通过局部线性模型近似复杂模型行为。这些方法已经被用于结合位点预测、免疫原性预测、稳定性分析以及抗病毒肽设计等任务。
此外,文章还讨论了counterfactual explanation(反事实解释)与contrastive explanation(对比解释)。反事实方法关注“最小改变”,即改变哪些残基可以导致输出发生变化;而对比解释则分析为何模型选择结果A而非结果B。
这些方法已经开始应用于AlphaFold等结构预测模型。例如,ExplainableFold能够识别哪些残基对于维持结构稳定性最重要,并预测哪些突变是“安全”或“危险”的。
文章还强调了生物安全问题。由于蛋白语言模型可能生成毒素或病毒相关序列,因此对模型鲁棒性与安全性的测试尤为重要。研究人员已经开始通过adversarial attack与prompt injection等方法测试模型漏洞,并分析训练数据泄漏问题。
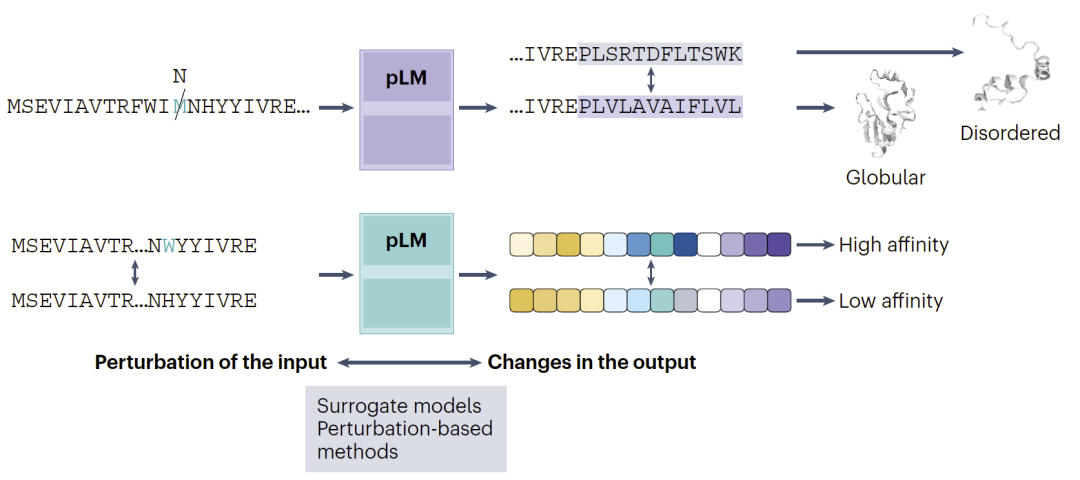
图5:输入扰动与输出变化分析。
XAI在蛋白研究中的五种角色
文章最核心的贡献之一,是提出了XAI在蛋白研究中的五种角色框架。
第一种角色是Evaluator。当前绝大多数XAI工作都属于这一类型,即验证模型是否学习到了研究人员预期中的规律。例如,若binding residue被赋予更高attention或gradient attribution,则说明模型确实学会了相关生物物理规律。
第二种角色是Multitasker,即将模型中发现的模式迁移到新的注释任务中。例如,将attention pattern用于新的binding residue预测。
第三种角色是Engineer。研究人员通过识别不重要的模块,对模型进行剪枝、压缩或架构优化,从而降低计算成本。
第四种角色是Coach。研究人员通过调控feature activation或修改模型权重,引导模型生成特定性质蛋白。例如,利用SAE增强与酶活性相关的latent feature,从而生成更高活性的酶。
最后一种角色则是Teacher,也是文章认为最重要、最具革命性的方向。Teacher意味着XAI能够帮助研究人员发现真正未知的新生物学规律,而不仅仅验证已有知识。例如,模型可能自动发现新的催化网络、折叠规律或进化模式。
然而,作者也强调,实现Teacher极具挑战性。因为模型可能只是学习统计偏差,而并未真正理解生物规律。因此,未来必须结合可靠的解释方法、严格benchmark以及湿实验验证,才能真正实现这一目标。
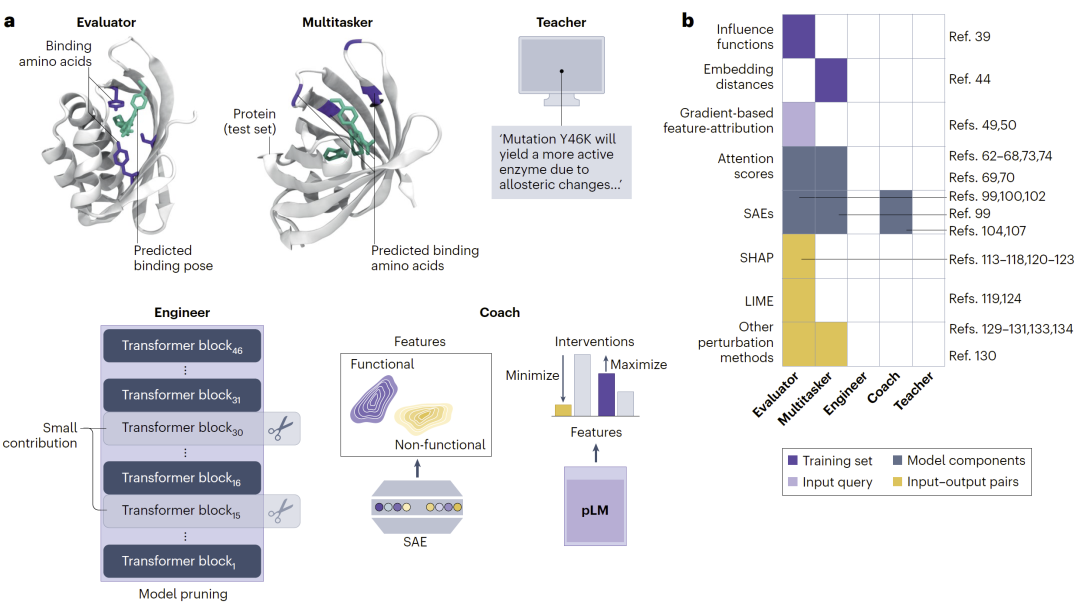
图6:XAI在蛋白研究中的五种角色框架。
讨论
文章认为,蛋白语言模型已经成为现代蛋白研究的核心工具,但其“黑箱化”问题正在成为制约进一步发展的关键瓶颈。XAI不仅能够提升模型透明性,还可能成为连接人工智能与生物学规律的重要桥梁。
当前,大部分研究仍停留在Evaluator阶段,即验证模型是否学习到了研究人员已经知道的规律。然而,真正具有变革性的方向,是利用XAI帮助研究人员发现未知规律,实现Teacher角色。这将使AI从“预测工具”转变为“科学发现伙伴”。
为了实现这一目标,研究人员需要建立更加可靠的benchmark、更高可信度的解释框架、更符合人类认知的可视化系统,以及更加系统的实验验证流程。同时,未来XAI方法也需要从NLP和计算机视觉领域进一步向生物学场景适配,发展真正面向蛋白与生命科学的解释框架。
文章最后强调,如果这些问题能够得到解决,那么未来XAI不仅能够提高蛋白预测与生成的可靠性,还可能帮助研究人员真正理解蛋白折叠、酶催化以及生命演化背后的基本原理。
整理 | DrugOne团队
参考资料
Hunklinger, A., Ferruz, N. Towards the explainability of protein language models. Nat Mach Intell (2026).
https://doi.org/10.1038/s42256-026-01232-w

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢