
如果把先导化合物优化想象成一场药物化学家的手工改造,过去很多工作像是在同一辆车上换轮胎、换车灯、换座椅:分子主体不大动,围着几个取代基反复试。问题是,药物研发真正难的地方往往不是把某一个指标调高,而是在活性、类药性、合成可及性、结构新颖性、血脑屏障通透性之间找到一个能站得住的平衡点。
最近,来自澳门理工大学应用科学学院的姚小軍和劉煥香老师团队发表在 Advanced Science 的一项研究,提出了一个名为 SMarT-Diff 的扩散生成模型,全称是 Scaffold-based Multi-property Tuning Diffusion,可以理解为一个带着药物化学经验做分子生成的模型。
它的核心不是单纯生成更多分子,而是把药物化学中非常重要的骨架跃迁策略融进扩散模型:保留能和靶点结合的关键药效团与空间关系,同时主动换掉分子的核心骨架,让模型走出原来化学空间的舒适区。
作者不只做了计算评估,还合成了 3 个模型生成的 LRRK2 候选分子并做了体外实验。其中 lrrk2_m_1001 对 LRRK2 的 IC50 达到 1.544 nM,优于阳性对照 LRRK2-IN-1 的 3.141 nM。对一个分子生成模型来说,这样的实验结果让它不再只是屏幕上的漂亮 docking 分数,而是有了真实药化验证的支点。
为什么药物生成模型不能只会生成分子
药物研发里的先导优化,本质上是一场多目标拉扯。一个候选分子活性更强,不代表它更像药;更像药,不代表它容易合成;容易合成,也不代表它能穿过血脑屏障;能穿过血脑屏障,又可能带来安全性和选择性的新问题。药物化学家每天面对的不是一道单选题,而是一张互相牵制的网。
传统先导优化通常从一个命中化合物或先导化合物出发,围绕结构做一轮又一轮修改。比如换一个芳环、加一个氟、调一个氢键受体、减少一个可旋转键,再回到实验台看活性、溶解度、代谢稳定性有没有变好。这个过程很有经验含量,但成本高、周期长,而且常常出现一种尴尬局面:某个指标变好了,另一个指标却掉下去了。药物发现并不是沿着一条直线前进,而是在高维化学空间里不断绕路。
AI辅助药物设计之所以被寄予厚望,是因为生成模型可以系统性地探索化学空间。扩散模型、流模型、大语言模型、强化学习、蒙特卡洛树搜索等方法都已经被用于分子生成和多目标优化。它们可以根据靶点口袋、理化性质或药效团约束,生成大量候选分子。
但这里有一个容易被忽略的问题:很多模型会生成新分子,却不一定懂药物化学里的骨架。
在药物化学中,分子骨架不是一个随便的形状。它决定了分子的拓扑结构、刚柔性、取代基伸展方向,也影响分子能否稳定占据结合口袋。所谓骨架跃迁,不是简单地把一个分子改得面目全非,而是在保留关键药效团和结合模式的基础上,换一个新的核心结构。它常被用于改善药代性质、绕开专利空间、提高选择性,或摆脱原始骨架带来的毒性和代谢风险。
这也是 SMarT-Diff 试图解决的问题:模型不能只会在已有分子附近做小修小补,也不能一味追求新颖而丢掉活性。它需要学会一种更像药化师的判断:哪里该保留,哪里可以换,换到什么程度才仍然像一个有希望的先导分子。
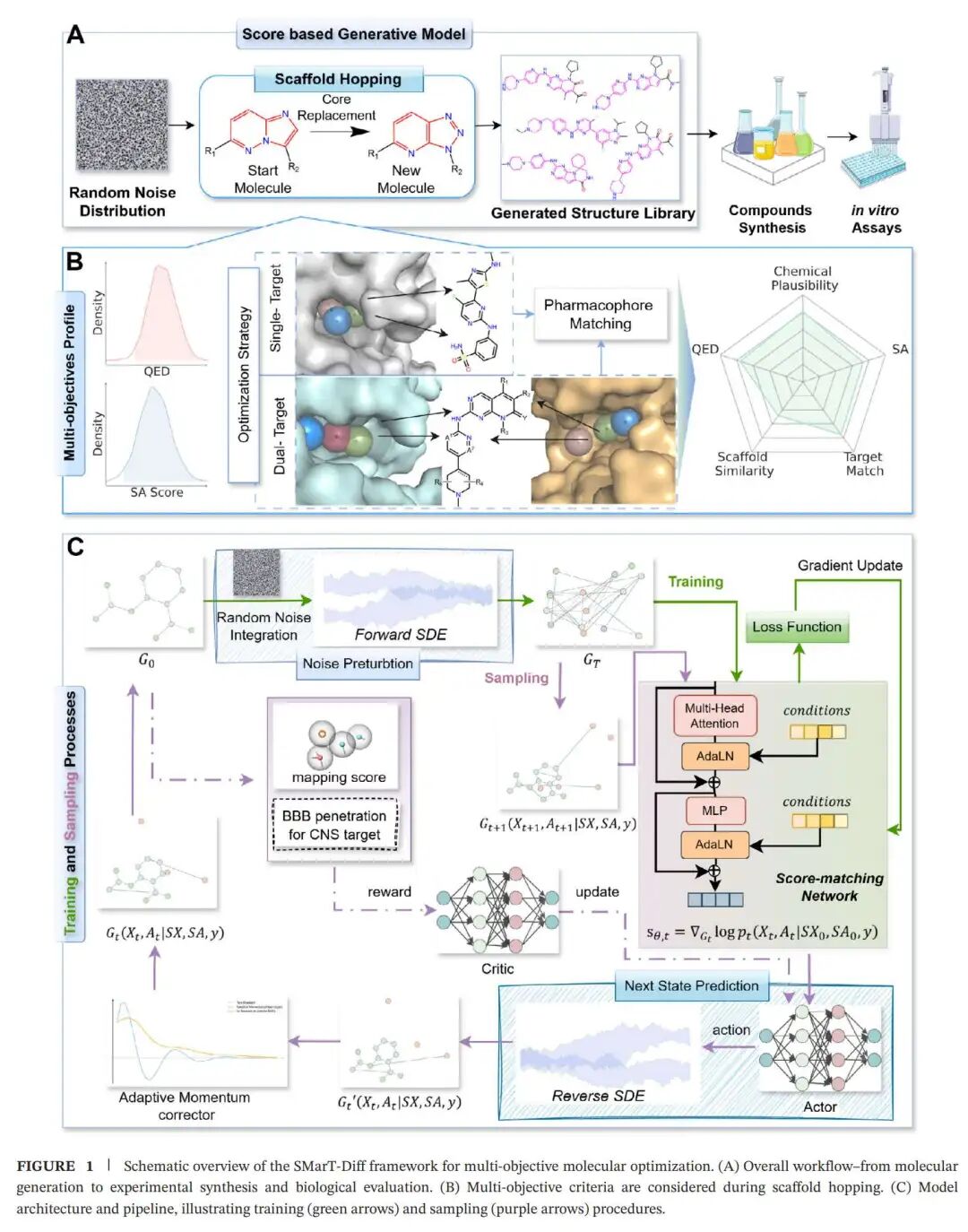
SMarT-Diff 做了什么:把骨架跃迁写进扩散模型
上图可以拆成三层来看。
第一层是分子生成的底座。作者使用的是基于得分的生成模型,也就是 score-based generative model。简单说,模型先把真实分子逐步加噪,直到它们变成接近随机噪声的状态;训练时再学习如何从噪声一步步还原出合理分子。这个框架适合处理连续的生成过程,也能和条件约束结合。
第二层是结构感知。SMarT-Diff 不是只把分子当成字符串,而是把分子表示成图结构:原子是节点,化学键是边。作者进一步引入图扩散 Transformer,让模型学习分子拓扑中的局部连接、边特征以及骨架与取代基之间的关系。对药物分子来说,这一点非常关键,因为同样的原子组成,连接方式不同,药效可能完全不同。
第三层是多目标引导。模型使用 Bemis-Murcko 骨架作为结构先验,同时纳入 QED、SA、药效团匹配等条件。QED 反映类药性,SA 反映合成可及性,药效团匹配则用于保留与靶点活性相关的关键空间特征。对于中枢神经系统相关靶点,模型还加入血脑屏障通透性预测,让候选分子不只是能结合靶点,也更接近中枢药物的现实需求。
这里有一个很重要的设计:SMarT-Diff 并不是让扩散模型单独完成所有事情。作者在采样阶段加入了 A2C 强化学习采样策略,也就是优势演员-评论家框架。可以把它理解成一个外层调参器:扩散模型负责生成结构,A2C 负责不断推动生成过程朝药效团匹配和目标性质更好的方向走。模型内部还有反向扩散预测器和自适应动量校正器,用来提升采样效率和稳定性。
这套设计背后的药化逻辑很清楚:先用骨架告诉模型不要乱走,再用性质指标告诉模型不要只顾活性,最后用药效团和强化学习把分子拉回靶点相关的方向。
新颖性不是越远越好,而是有控制地离开原始骨架
分子生成领域常常会强调 novelty,也就是新颖性。但对药物设计来说,太新有时反而危险。一个分子如果和已知活性分子完全没有结构关系,确实新,但它可能也失去了已知药效团和结合模式。反过来,如果生成分子和参考分子太像,就只是类似物扩展,谈不上真正的骨架跃迁。
SMarT-Diff 的创新点就在这个中间地带:它试图把生成分子控制在一个合理的骨架跃迁区间。
作者在评价中使用了 Bemis-Murcko 骨架相似性。论文里将 0.3–0.4 附近视为较理想的骨架跃迁区间:相似度太高,说明模型还停留在类似物生成;相似度太低,可能已经丢掉了关键药效关系。SMarT-Diff 在 LRRK2 基准任务中取得的骨架相似度为 0.362,同时保持较高结构多样性,这说明它并不是通过简单复制参考分子来获得好结果。
消融实验也支持这一点。单纯的生成器能生成有效分子,但靶点相关性不足;引入骨架条件后,模型回到更合理的药物化学空间;加入 RA 采样后,骨架相似性进一步提升,但也更容易生成类似物;再加入 A2C 外层优化后,模型把骨架相似度压回更接近骨架跃迁的区域,同时维持类药性、合成可及性和预测结合能力之间的平衡。
换句话说,这个模型的价值不是更会生成,而是更会把生成限制在有药化意义的范围里。
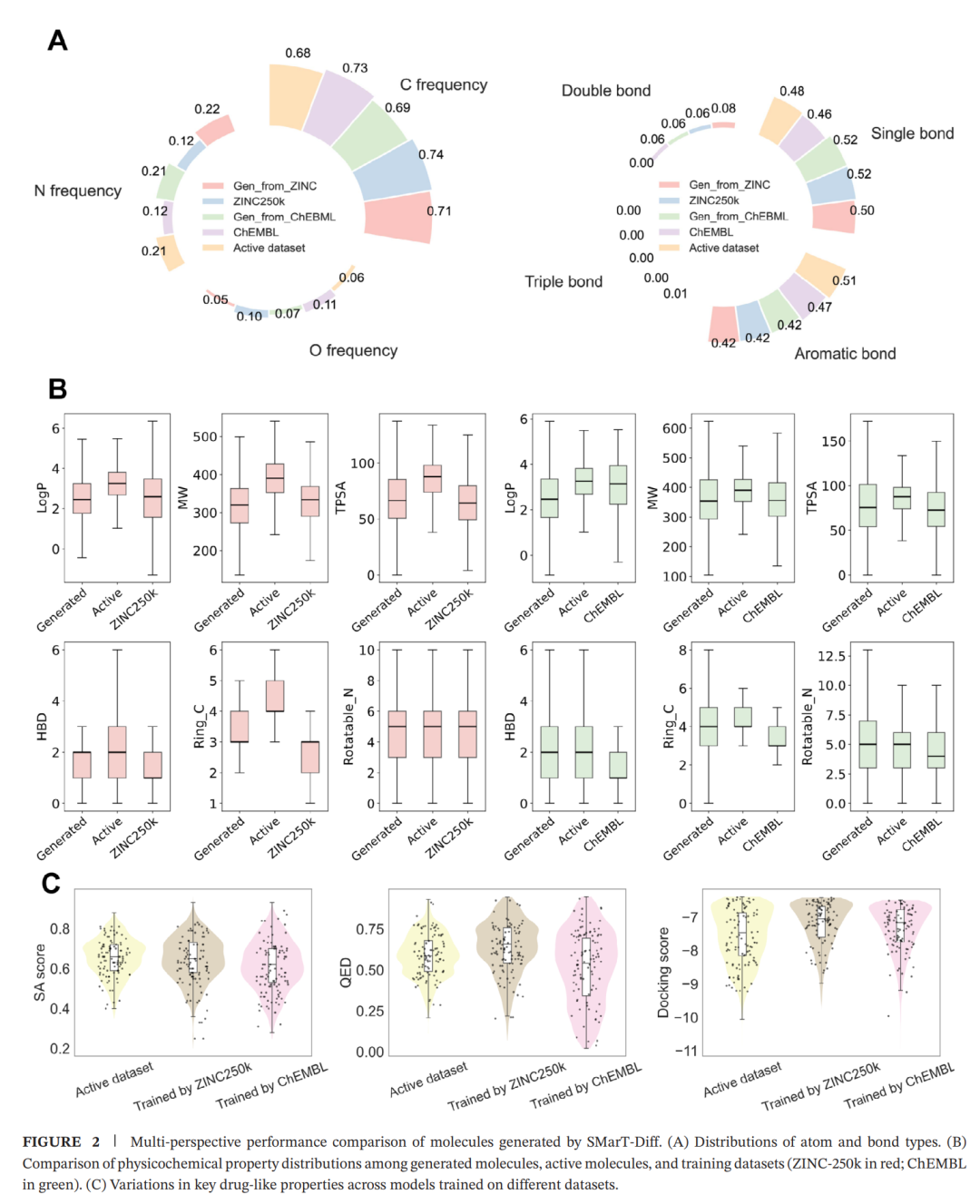
实验结果一:模型不是简单背数据,而是在不同化学空间中重新校准
作者分别使用 ChEMBL 和 ZINC-250k 训练模型,并在 LRRK2 任务中比较生成分子的表现。这个实验的意义在于检验模型是不是只会记住某一个数据集的统计分布。
从图2可以看到,生成分子的原子类型、键类型和理化性质分布会受到训练集影响,这很正常。ChEMBL 本身更偏向已知生物活性分子,ZINC-250k 更偏向可购买或药物样小分子库,两个数据集的化学分布不同,模型学到的先验也不同。
更关键的是,SMarT-Diff 生成的分子并没有机械地停留在训练集分布中,而是在训练集和活性参考分子之间形成过渡。比如脂溶性、分子量、极性表面积、氢键供受体数、可旋转键等指标,整体上落在更接近药物样分子的区域。对于 LRRK2 这样的中枢相关靶点,血脑屏障通透性约束也让模型更注意疏水性和极性之间的平衡。
这说明 SMarT-Diff 的生成不是盲目扩散,而是一种带有任务方向的化学空间迁移:从训练集学到可行分子的基本语法,再通过药效团和多性质条件,把生成结果推向更接近目标活性的区域。
实验结果二:真正的骨架外推,不能牺牲结合能力
作者进一步看了 SMarT-Diff 在 LRRK2 上的骨架级别外推能力。这里的评估很有意思:他们不仅看 Bemis-Murcko 骨架,还看更严格的通用骨架,也就是把所有原子简化成碳、所有键简化成单键后的拓扑框架。后者更能检验模型是否真的生成了新的拓扑结构,而不只是做了一些生物电子等排替换。
在针对 LRRK2 生成的 10000 个分子中,93.96% 的分子具有训练集中不存在的 Bemis-Murcko 骨架,60.08% 具有训练集中不存在的通用骨架。与参考活性分子相比,新颖性也分别达到 81.87% 和 60.20%。这说明模型确实跳出了已知骨架。
但更重要的是,跳出去之后没有明显丢掉结合潜力。在经过 drug-likeness 过滤的子集中,也就是 docking 分数低于 -8.0 kcal/mol、QED 高于 0.6、SA 高于 0.6 的分子里,那些最近邻相似度低于 0.5 的分子仍然取得了 -8.57 kcal/mol 的中位 Glide SP docking 分数。这意味着模型在探索新骨架时,仍能保留一定靶点相关的结合特征。
这个结果对药物设计很关键。因为骨架跃迁最怕的不是换不掉骨架,而是换掉以后只剩下形式上的新颖。SMarT-Diff 至少在计算层面证明,它能把新颖性和结合潜力同时保住。
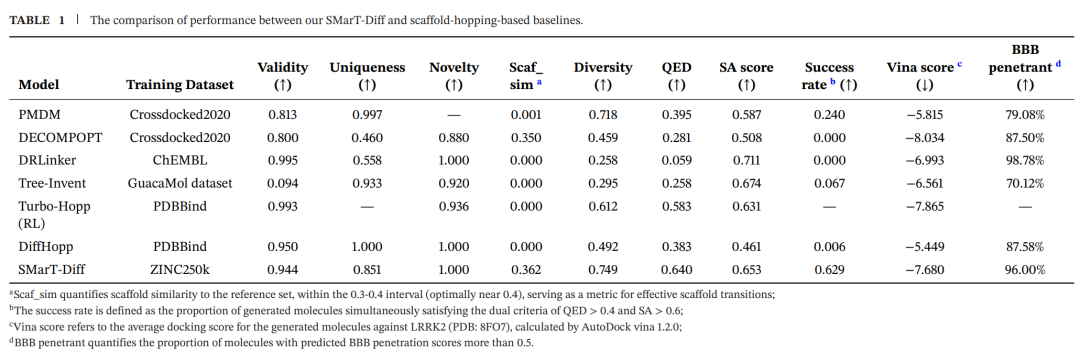
实验结果三:和基线模型相比,SMarT-Diff 赢在平衡
上表是整篇文章中最适合细读的一张表。作者把 SMarT-Diff 与 PMDM、DECOMPOPT、DRLinker、Tree-Invent、TurboHopp、DiffHopp 等骨架跃迁或结构生成相关模型进行了比较。
单看某一个指标,其他模型并非没有亮点。比如 DRLinker 的 SA 分数较高,TurboHopp 和 DECOMPOPT 的预测结合分数也很强。但药物设计不看单项冠军,而看候选分子能不能同时满足多个条件。
SMarT-Diff 在 LRRK2 任务中表现出几个值得注意的数字:
有效性 0.944,唯一性 0.851,新颖性 1.000,骨架相似度 0.362,多样性 0.749,QED 0.640,SA 0.653,成功率 0.629,Vina 分数 -7.680,预测血脑屏障通透比例 96.00%。
这里的成功率定义为生成分子同时满足 QED > 0.4 和 SA > 0.6。SMarT-Diff 的成功率明显高于最接近的基线 PMDM 的 0.240。更重要的是,SMarT-Diff 没有通过把分子生成得极其接近参考分子来提高成功率,它的骨架相似度落在较合理的跃迁区间,同时多样性仍然较高。
这就是这篇文章的核心判断:SMarT-Diff 的优势不在于某个指标压倒所有方法,而在于它把多个药化目标压在了同一张桌子上,并给出了一组更均衡的解。
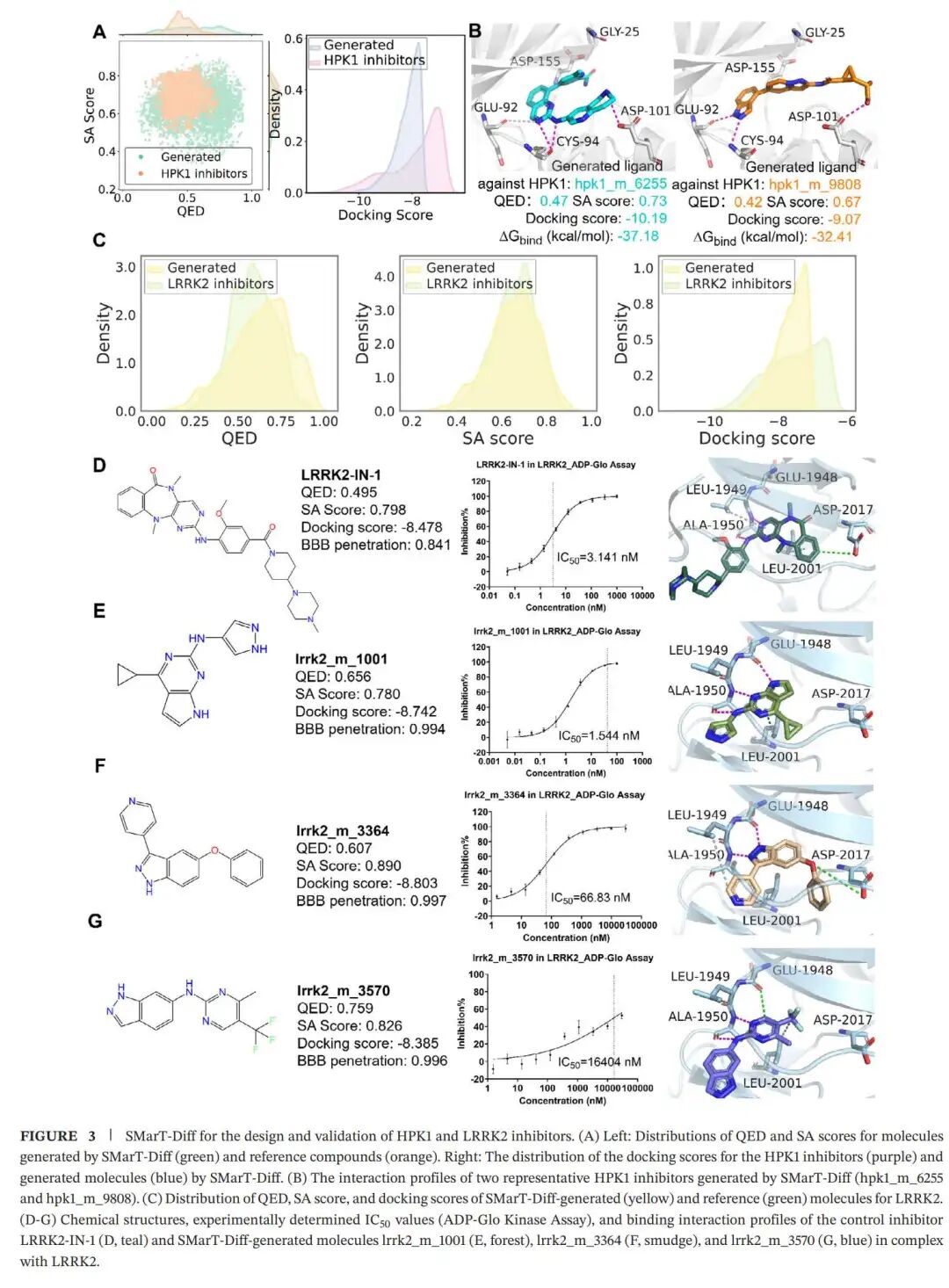
实验结果四:从计算结果走到湿实验,LRRK2 给了一个强支点
在单靶点任务中,作者选择了两个激酶靶点 HPK1、LRRK2,以及一个 B 类 GPCR 靶点 GLP-1R。
HPK1 部分主要是计算验证。生成分子的 QED 和 SA 分布与参考抑制剂相近,docking 分数集中在 -8.0 kcal/mol 附近。两个代表性分子 hpk1_m_6255 和 hpk1_m_9808 与 HPK1 的结合模式较相似,能与 Cys94 形成关键氢键,并通过水介导与 Asp155 形成氢键;其中 hpk1_m_9808 的吡唑环还与 Glu92 形成额外氢键。这些相互作用与已知 HPK1 抑制剂中的关键结合特征相符。
LRRK2 部分则更有说服力,因为作者做了合成与体外活性实验。研究团队从生成结果中选出 3 个代表性分子:lrrk2_m_1001、lrrk2_m_3364、lrrk2_m_3570。这些分子由商业实验室合成,并通过 ^1H-NMR 和 LC-MS 确认结构。随后作者使用 ADP-Glo kinase assay 检测它们对 LRRK2 及其 G2019S 突变体的抑制活性。
主文图3中给出的 LRRK2 IC50 结果非常清楚:
lrrk2_m_1001:1.544 nM
lrrk2_m_3364:66.83 nM
lrrk2_m_3570:16.404 μM
阳性对照 LRRK2-IN-1:3.141 nM
其中 lrrk2_m_1001 的活性优于阳性对照,这是整篇文章最有分量的实验结果。它说明 SMarT-Diff 生成的候选分子并非只是在计算评分上好看,至少有一个分子在真实体外酶活实验中表现出了很强的抑制能力。
从结合模式看,三个生成分子都能稳定占据 LRRK2 的 ATP 结合口袋。它们核心骨架中的吡唑环与铰链区残基 Ala1950 形成氢键,这通常是 LRRK2 抑制剂活性中的关键相互作用之一。lrrk2_m_1001 和 lrrk2_m_3364 还与 Leu1948 形成氢键,lrrk2_m_3570 则与 Leu1948 存在电性相互作用。Asp2017 以及 Leu2001、Leu1949 等周围残基也参与稳定结合。
这组结果给人的感觉很直接:模型生成的分子不仅像药,而且在关键结合口袋里做对了一些药化上重要的事。
GLP-1R 的测试则展示了跨靶点泛化能力。这个靶点属于 B 类 GPCR,正构口袋更大、更暴露,和激酶口袋差异明显。作者在不重新训练模型的情况下进行测试,生成分子的 QED 中位数达到 0.575,高于参考活性分子的 0.430;SA 中位数为 0.61,与参考分子的 0.69 接近。虽然整体 docking 分数中位数略弱于参考活性分子,但排名靠前的 6 个分子达到 -9.46 到 -11.45 的 docking 分数,其中 3 个低于 -10.0,并保持较高 QED。这说明模型并不是只适合激酶类靶点,但 GLP-1R 部分目前仍主要停留在计算验证层面。
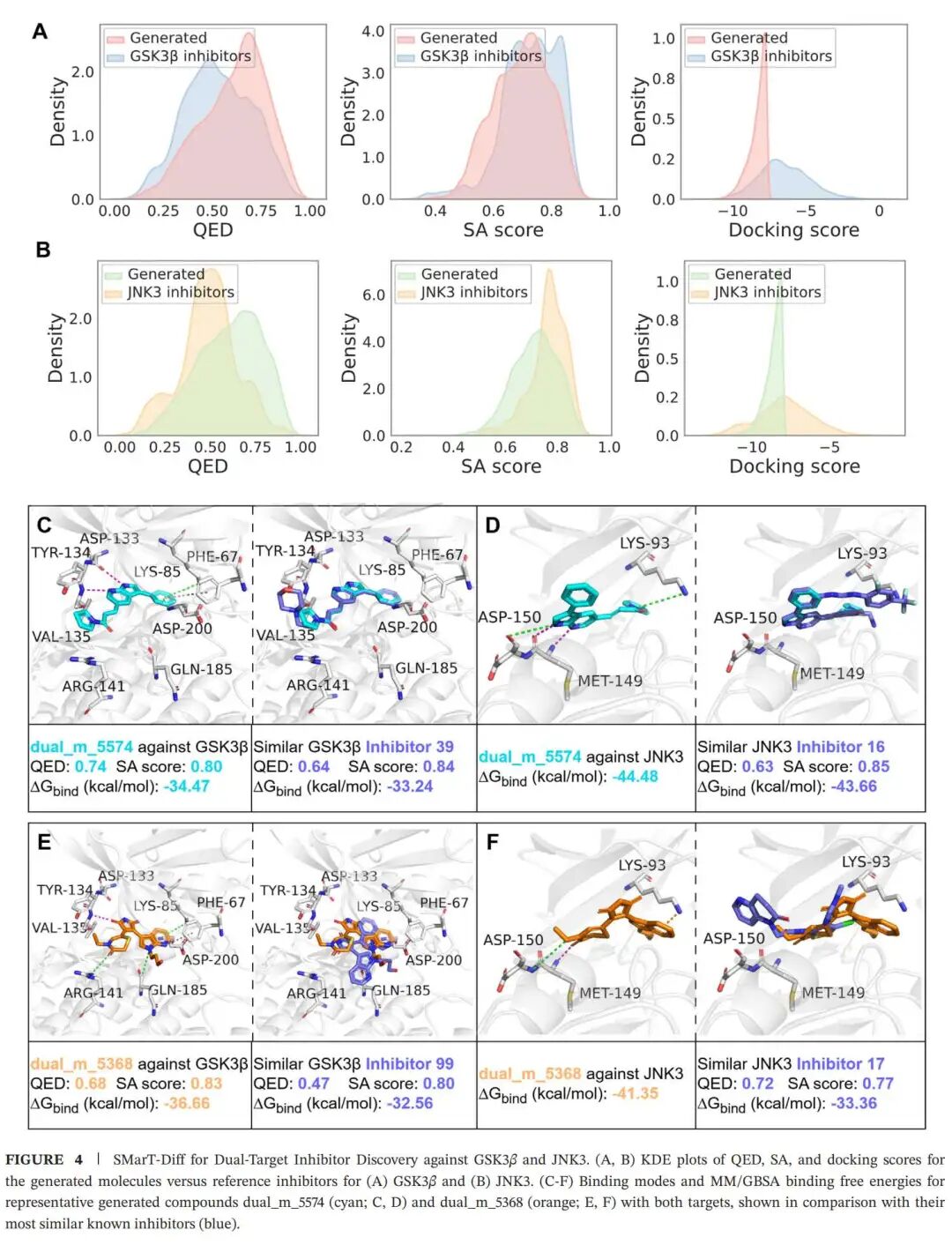
实验结果五:双靶点设计里,模型尝试绕开硬拼药效团的老办法
双靶点药物设计在复杂疾病中很有吸引力,比如癌症、自身免疫疾病以及神经退行性疾病。一个分子如果能同时调控两个相关靶点,理论上可能带来更好的疗效或更系统的通路干预。但这件事很难,因为两个靶点的结合口袋几何形状、疏水性、氢键网络可能都不一样。传统做法常常是把两个靶点的药效团硬拼在一起,结果分子变大、变复杂、合成更难,药代性质也容易变差。
SMarT-Diff 的双靶点策略更柔和一些。作者从已知 GSK3β 和 JNK3 抑制剂中提取 Bemis-Murcko 骨架,再通过最大公共子结构挖掘两类抑制剂共享的核心片段,最后选择覆盖这些共享核心的参考骨架来引导生成。这样做不是把两个药效团机械缝合,而是让模型在共享结构基础上重新组装可行分子。
在 GSK3β/JNK3 双靶点任务中,生成分子的 QED 大多落在 0.25–0.9,SA 大多落在 0.5–0.9,与参考抑制剂接近。docking 分数多数低于 -7.5 kcal/mol,并在 -8.5 kcal/mol 附近形成明显峰值。作者还从结果中筛选出双靶点表现较好的候选分子,用 MM/GBSA 估算结合自由能,发现部分生成分子的 ΔGbind 优于共晶配体。
两个代表性分子 dual_m_5574 和 dual_m_5368 在 GSK3β 和 JNK3 口袋中都能形成较稳定的氢键、电性和疏水相互作用,并与相似参考抑制剂的结合模式接近。这说明模型在双靶点场景下并不是只追求其中一个靶点的好分数,而是在尝试找到两个口袋都能接受的结构折中。
当然,这部分仍然是计算层面的发现,还没有像 LRRK2 那样进入合成和体外验证。但作为模型能力展示,它说明 SMarT-Diff 不局限于单靶点先导优化,也可以扩展到更复杂的多靶点分子设计任务。
把药化经验变成生成模型的约束语言
很多 AI 药物设计模型的问题,是生成结果和药物化学之间隔着一层。模型能给出分子,却很难解释为什么这个分子值得合成;模型能优化 docking 分数,却未必知道药物化学家最在意的是哪个骨架、哪个相互作用、哪个性质红线。
SMarT-Diff 的方向更接近药化实践。它没有把骨架跃迁当作一个事后分析指标,而是在生成过程中就把骨架、药效团、类药性、合成可及性和血脑屏障通透性一起纳入。这样生成出来的分子不是孤立的分数最优解,而是更接近先导优化中的候选方案。
尤其是 LRRK2 的实验验证,让这项工作从模型论文向药物发现应用迈了一步。lrrk2_m_1001 的纳摩尔级 IC50 说明,模型确实有可能在已知活性空间之外找到强活性分子。对生成式药物设计来说,哪怕只合成了 3 个分子,这个结果也比单纯展示上万个虚拟分子更有说服力。
END:AI 药物设计正在从会生成走向会取舍
SMarT-Diff 给出的启发是,药物生成模型的下一步不是单纯生成更多分子,而是学会药物化学中的取舍。哪些结构可以换,哪些药效团必须保留;哪些性质可以牺牲一点,哪些指标不能越线;怎样在新颖性和可验证性之间找到一个适合合成的候选分子。
这篇文章把骨架跃迁、多目标优化、扩散模型、强化学习采样和湿实验验证放到同一个框架里。它没有把 AI 描述成替代药化师的黑箱,而是让模型更接近药化师的工作方式:先理解已知活性分子的核心,再在可合成、像药、有希望结合靶点的空间里,提出新的结构方案。
如果说过去很多生成模型像是在化学空间里随机开灯,那么 SMarT-Diff 更像是拿着药化地图往前走。它未必每一步都能到达新药,但至少开始懂得,真正有价值的分子不是最陌生的那个,而是换了骨架之后,仍然能在靶点口袋里站稳的那个。
参考文献
Y.Yang, X.Gong, S.Gu, et al. “Diffusion-Based Generative Model With Scaffold-Hopping Strategy Yields Highly Potent Bioactive Molecules.” Advanced Science (2026): e75674.
https://doi.org/10.1002/advs.75674
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢