

期刊|Journal of Medicinal Chemistry(2026)
DOI|10.1021/acs.jmedchem.6c00537
单位|美国国立卫生研究院(NIH)国家转化科学促进中心(NCATS);合作单位 Frederick 国家癌症研究实验室蛋白表达实验室
一、摘要速览
本文提出并验证了一套以 AI 为引导、以化学反应为基础的 hit-to-lead 工作流,作者命名为 CSAR(Comprehensive Structure–Activity Relationship,全面构效关系)。其核心是把经典药物化学"逐位优化(R1 → R2)"的逻辑,通过 in silico 反应枚举放大到数十万分子规模,再以药效团对接与深度学习 QSAR(DLCA)进行双重三联打分与优先级排序,从而在保证可合成性的前提下系统性地扩展构效关系。
作者以醛脱氢酶 3A1(ALDH3A1)——一个与肿瘤、神经退行性疾病相关但被探索不足、且家族内选择性极难实现的靶点——作为验证案例。两轮枚举(R1 哌嗪 + R2 酰胺)共生成约 25 万个可合成虚拟类似物,经计算分流后仅合成 150 个化合物,即把生化活性从苗头的 1.41 μM 提升到约 1 nM(NCATS-SM0707,提升约 1000 倍),并获得 4 nM 级细胞活性(NCATS-SM0708)。两个先导物对 ALDH 家族其它同工酶均表现出优异选择性,且经底物竞争实验与双吖丙啶光亲和探针 + 质谱证实其结合于 ALDH3A1 经典底物口袋。
二、研究背景与立题动机
2.1 真正的瓶颈在 苗头到先导
尽管高通量筛选(HTS)、结构建模与机器学习已显著加速早期苗头识别,但从初始苗头推进到兼具高活性、高选择性与可接受成药性的先导化合物,仍需大量时间与资源,对于化学可处理性有限的靶点尤其如此。这一阶段才是多数项目真正的耗时点。
2.2 传统优化的天花板
经典的逐步 R 基团优化与 Topliss 取代策略逻辑清晰、可解释,但本质上受限于有限的商业砌块与"按目录取材(analog-by-catalog)"的取向,SAR 探索往往被困在熟悉的化学空间内。靶点越复杂、对选择性要求越高,这种小尺度、串行式优化越难触及发现高质量新化学实体所需的广阔空间。
2.3 现有生成式平台的短板
作者明确把本工作与既有平台做了区隔:REINVENT(强化学习引导分子生成)、MegaSyn(整合合成可行性评分)、AutoSynRoute(逆合成路线规划)等在骨架跃迁与点子生成上很强,但它们多为早期探索而设计,并非针对特定骨架的 SAR 精细扩展。其通用反应规则常缺乏微调骨架所需的化学语境——而对 ALDH3A1 这类"取代基微小变化即引发活性或同工酶选择性剧变"的靶点,这恰恰是关键短板。CSAR 正是为弥合这一空档而生:它既保留经典药化的逐位优化逻辑,又借助经过精心设计的反应模板实现 in silico 的规模化。
2.4 为何选择 ALDH3A1 作为验证靶点
ALDH3A1 属 NAD(P)⁺ 依赖的醛脱氢酶家族,负责解毒脂质来源醛类,牵涉氧化还原调控、化疗耐药与免疫逃逸,并在肺癌、乳腺癌、头颈癌等实体瘤中高表达、与不良预后相关。它"价值明确却工具匮乏":既往强效且选择性的小分子抑制剂稀少(如 CB7 等),且 ALDH 家族成员序列与活性口袋高度同源(其中 ALDH3A2 与 ALDH3A1 同源约 70%),使家族内选择性成为公认难题。一个"又难又有意义"的靶点,正适合检验新方法的真实价值。
三、方法学:CSAR 平台拆解
3.1 总体架构与设计哲学
CSAR 的内核可概括为一句话:沿用经典 R1 → R2 逐位优化的逻辑,但用算力放大上千倍,并在每一步都嵌入"可合成性"约束。 整条流水线由三个可独立替换、可叠加的模块组成,且框架本身并不局限于本文演示的两个反应/两个位点,原则上可通过追加反应方案(如 linker 修饰、骨架多样化)与迭代设计周期进行扩展。
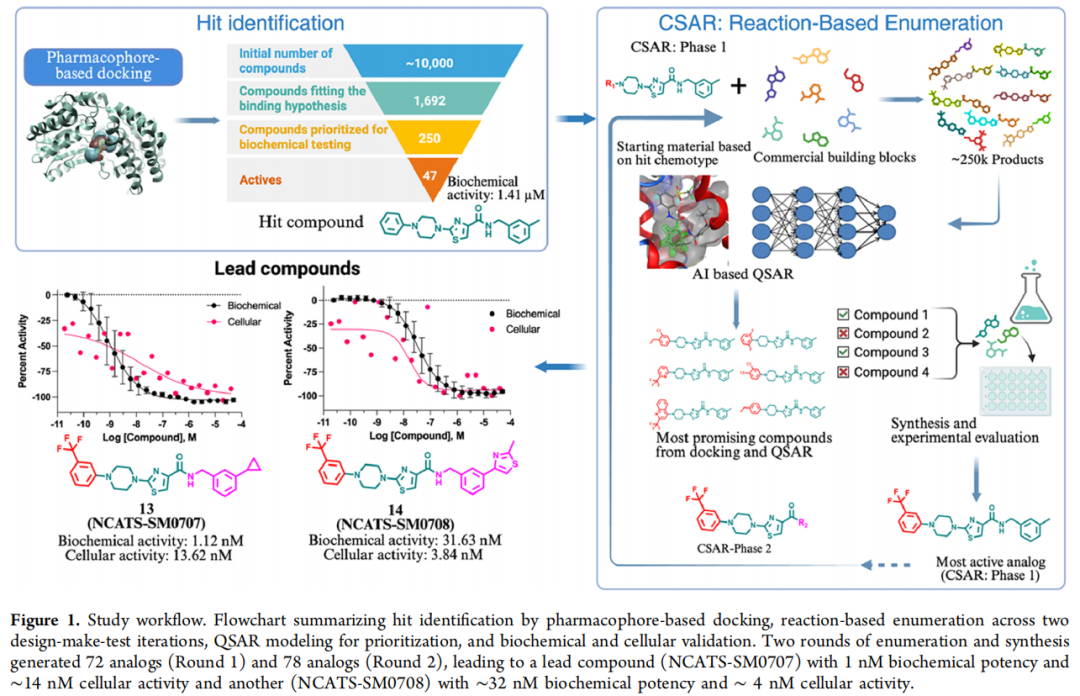
3.2 模块一:药效团模型与结构导向虚拟筛选
基于人源 ALDH3A1 晶体结构(PDB ID: 4H80),在 MOE(Molecular Operating Environment) 中构建结构化药效团模型,捕捉底物结合通道内的关键作用特征:氢键供体/受体、疏水区域,以及邻近辅因子结合位点的芳香核。蛋白预处理包括去水与无关杂原子、加氢、指认质子化状态、能量最小化。对接采用 MOE 的 Affinity dG 打分函数,对约 10,000 个结构多样化合物(NCATS 内部库,经 3D 构象生成与质子化预处理)进行评分与排序。
3.3 模块二:反应枚举(in silico synthesis)
枚举在 KNIME 平台中借助 RDKit 双组分反应节点(two-component reaction) 实现:先编写反应 SMARTS,再将选定骨架与 Enamine 商业砌块组合,系统生成可合成虚拟库。两个反应模板分别针对骨架两侧:
• R1 位 — 芳香亲核取代(SNAr):以 2-氯噻唑中间体为底物,与超过一百万个 N-取代哌嗪砌块虚拟反应,生成 9,336 个类似物(变异严格限定在哌嗪 N 上的取代基)。实际合成条件:DMSO,110 °C,6 h(通用流程 A)。 • R2 位 — 酰胺偶联:以噻唑-4-甲酸中间体与商业一级胺虚拟偶联,生成约 251,225 个类似物。实际合成条件:N-甲基咪唑、TCFH、MeCN,室温,30 min(通用流程 B)。
一处可留意的术语细节:正文讨论部分一处将骨架 A 称为"thiazole-containing sulfonamide scaffold(含磺酰胺的噻唑骨架)",但表 1、表 2/3 与反应方案显示真实化学为 2-(哌嗪-1-基)噻唑-4-甲酰胺(噻唑-甲酰胺核心,2-位连 N-取代哌嗪),并非磺酰胺。这属文中用词不一致,解读时应以实际结构为准。
3.4 模块三:QSAR 建模与化合物优先级
预测打分采用作者团队此前发表的深度学习共识架构(DLCA, Deep Learning Consensus Architecture):它整合多组在不同分子表征上训练的深度神经网络——Morgan、Avalon、AtomPair 指纹、RDKit 物化描述符,以及一个基于 SMILES 字符串的卷积神经网络——通过对各网络输出取平均得到共识分数,在兼顾各表征优势的同时降低误差传播。作者同时构建了基于上述指纹/描述符及其组合的随机森林(RF)基线作对照。
可靠性方面有两点工程细节值得称道:其一,采用基于 Morgan 指纹 Tanimoto 相似度(阈值 ≥ 0.6) 的适用域(applicability domain)过滤,将预测约束在与已知活性物相近的化学空间内,降低外推不确定性;其二,回归任务中执行了批平衡(batch balancing),以消除模型偏向低活性化合物的预测偏倚。
| 0.51 | ||
| 0.74 | ||
R² = 0.51 已"足以指导早期选型";第二轮纳入 R1 真实数据重训练后提升至 0.74,在新枚举化学空间内的排序分辨力更佳——这本身也展示了 design-make-test 循环中数据反哺模型的价值。
四、结果
4.1 苗头识别与骨架选择
虚拟筛选呈典型漏斗式收敛,并通过"近似物挖掘"扩大命中:
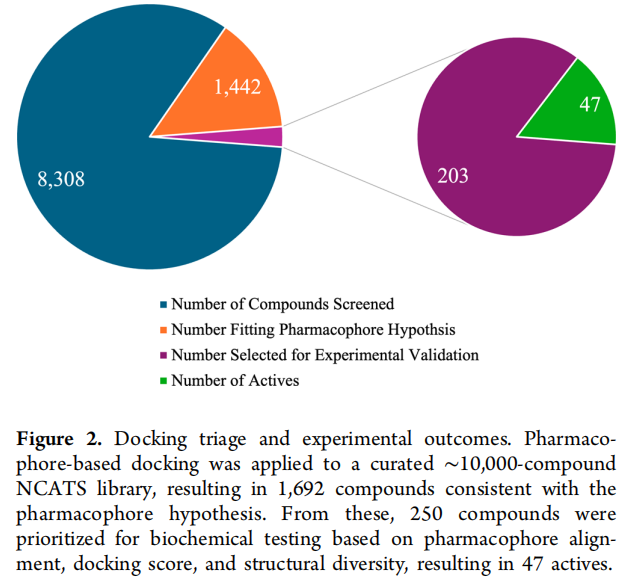
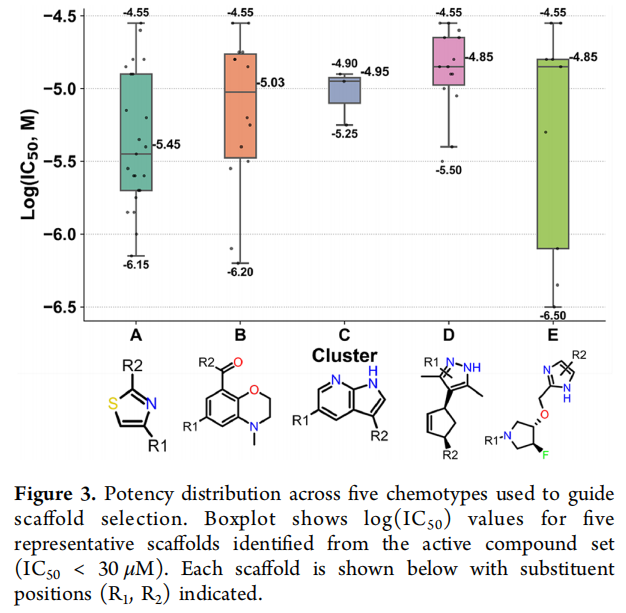
骨架 A(含噻唑)入选的理由是综合性的:一致的中等微摩尔活性、良好的早期 ADME、清晰的药效团契合,以及适于进一步修饰的功能化把手;其平面芳香核与均衡的物化性质提供了可处理的优化起点。库中检索到 48 个 A 的类似物,其中 43 个共享 N-取代哌嗪 + 酰胺(对应 R1/R2)的共同骨架,23 个表现出可测活性(IC₅₀ 0.7–28 μM,效价 33%–92%)。早期 SAR 已显示:R2 位取代苄胺远优于杂芳胺与脂肪胺,二级酰胺活性下降;而 R1 位库内仅有单取代 N-苯基哌嗪、信息有限——这恰恰为后续反应枚举指明了扩展方向。原始苗头活性 IC₅₀ = 1.41 μM(效价 74%)。
4.2 第一轮(CSAR Phase I):R1 哌嗪枚举
哌嗪母核及其取代模式经对接与 QSAR 评估被保留(得分最优),R1 作为不破坏核心骨架与关键药效团接触的可及把手。9,336 个枚举分子经 DLCA 打分 + 药效团对接,筛出 500 个进入终审,与药化专家共同评估可合成性与 SAR 相关性后选定 80 个砌块,最终成功合成 72 个(成功率 ≈ 90%)。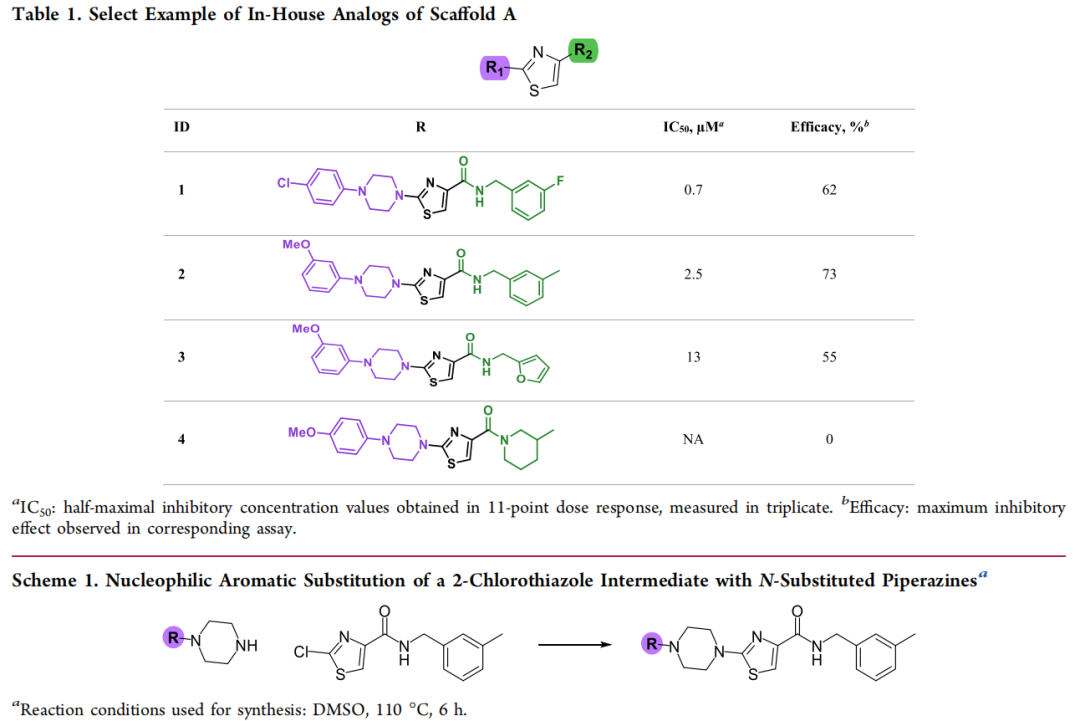
生化结果亮眼:70/72(97%) IC₅₀ < 30 μM 且效价 > 30%,多数落在 0.23–20 μM,其中 48 个优于原始苗头。SAR 上,取代苯基哌嗪 + 较小取代基最受偏好;值得注意的是吡啶基哌嗪(化合物 11) 在不牺牲活性的前提下显著改善溶解度——这是一条有价值的物化优化线索。细胞层面采用 ALDEFLUOR 高内涵成像,以优先表达 ALDH3A1 的 OE19 细胞系评估:53/72(73%) 细胞 IC₅₀ < 10 μM、效价 > 50%。综合生化与细胞活性,化合物 5(229 nM 生化 / 3.13 μM 细胞) 被选为第二轮优化起点。
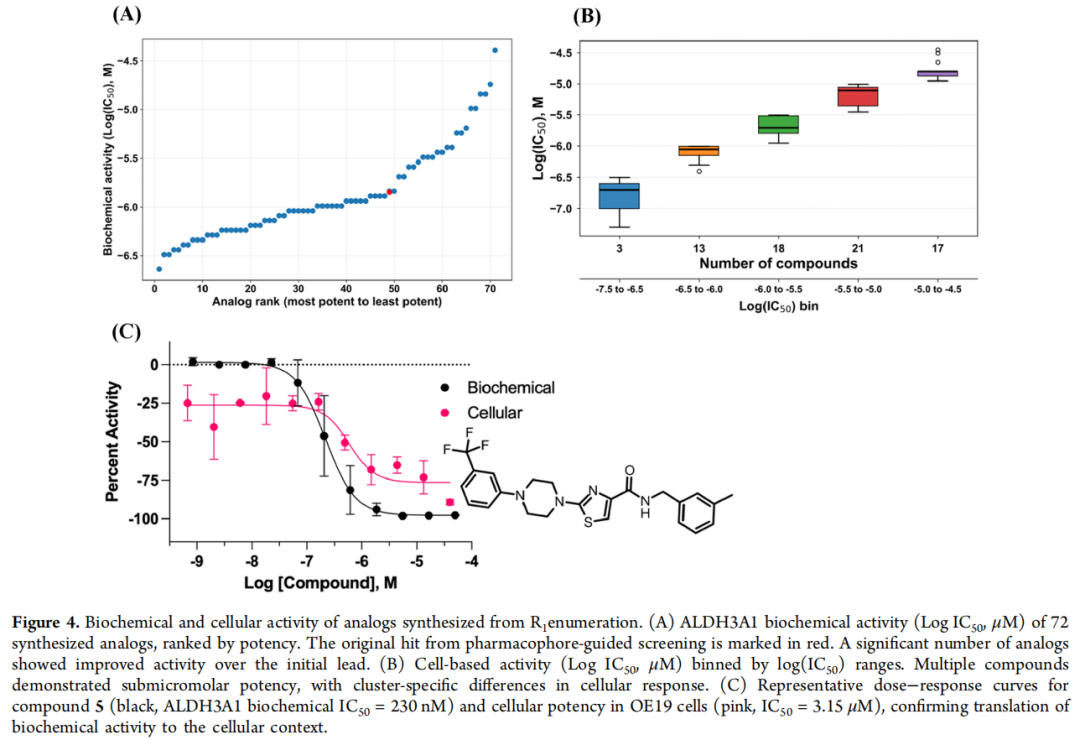
4.3 第二轮(CSAR Phase II):R2 酰胺枚举
以化合物 5 为基础,对 R2 位酰胺进行枚举:251,225 个分子经重训练 QSAR(R² = 0.74) 初筛出 40,664 个,再经药效团对接收敛至 top 700,与药化专家结合预测分、砌块成本与到货周期选定 85 个,成功合成 78 个。
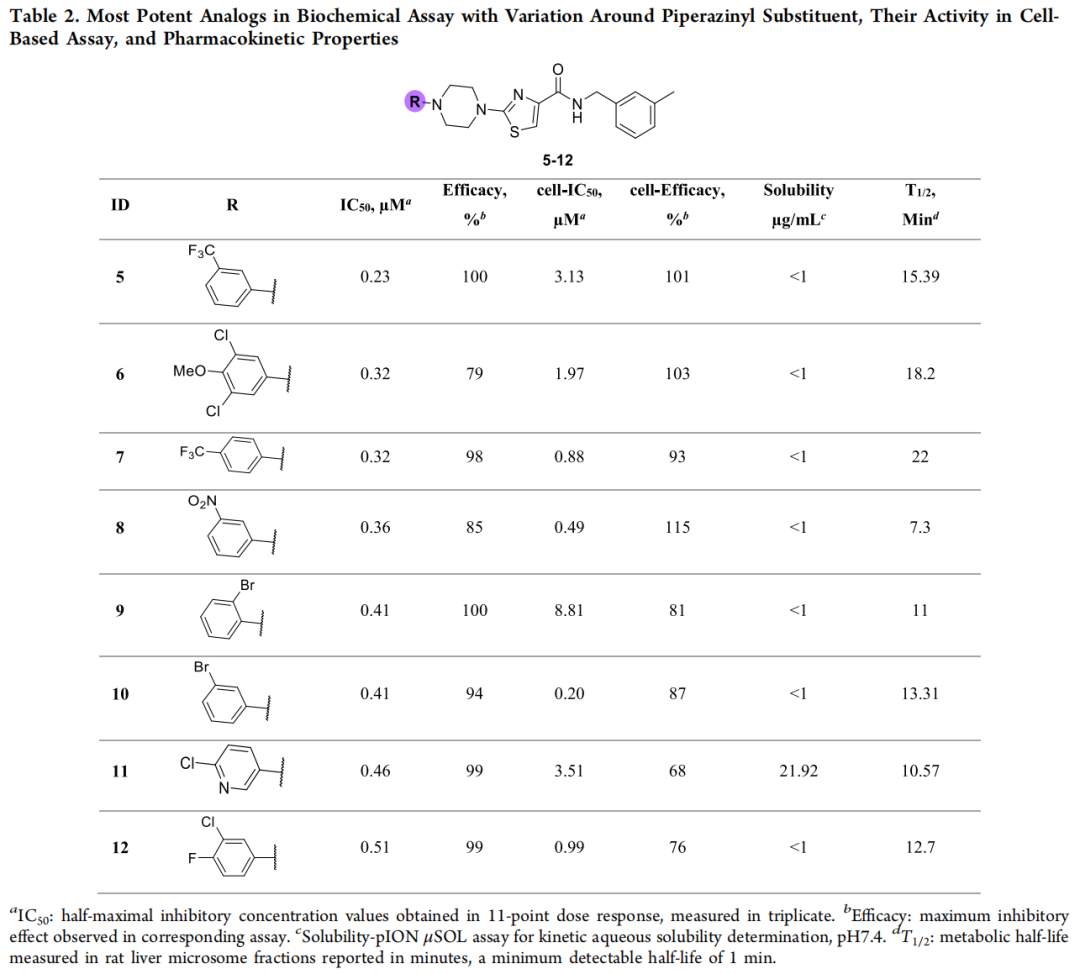
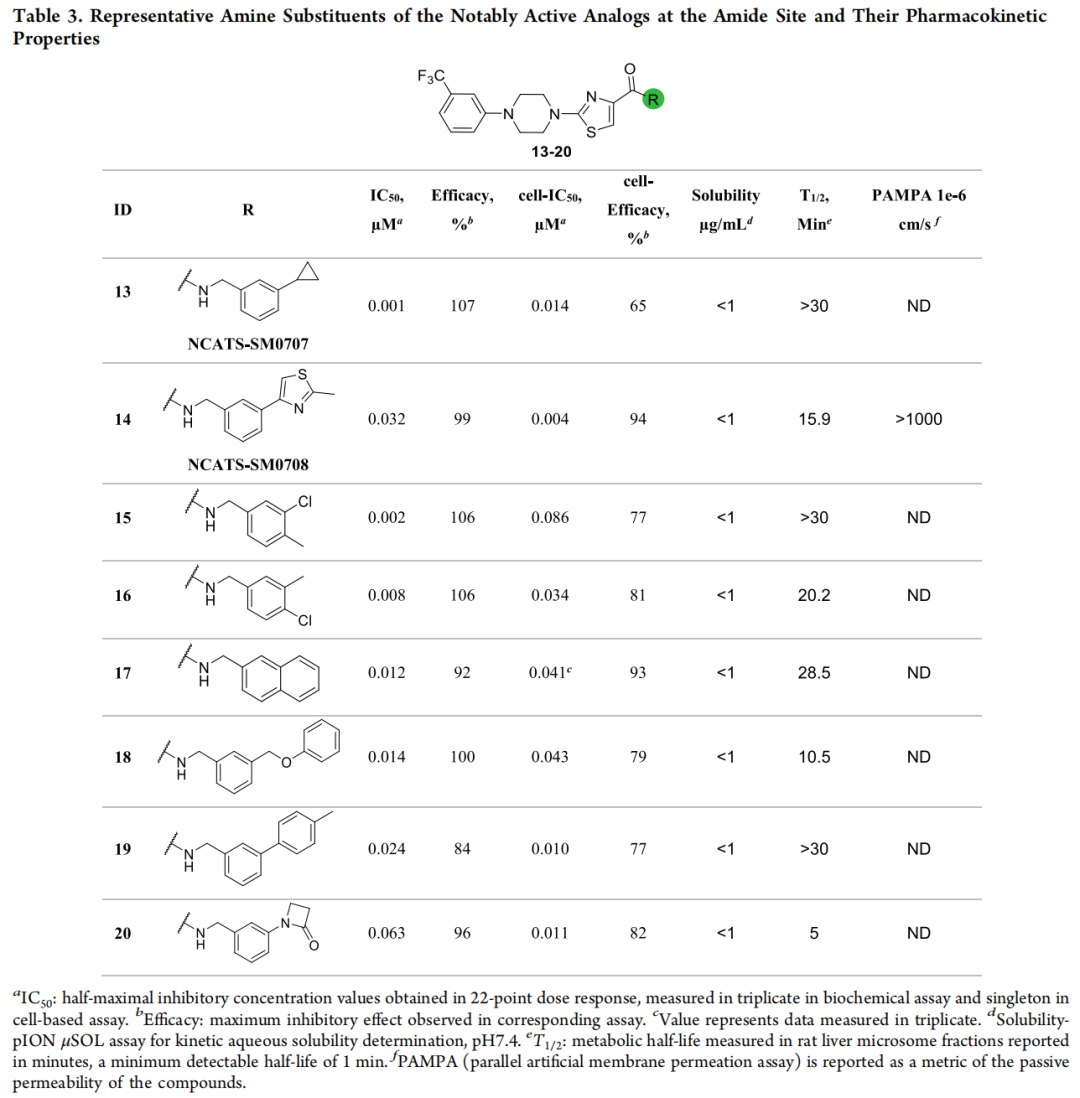
生化上,72/78(92%) IC₅₀ < 20 μM,其中 8 个进入个位数 nM;以化合物 5(229 nM)为基准,43 个 R2 类似物更强。细胞活性提升尤为显著——57 个细胞 IC₅₀ < 10 μM、多个达亚微摩尔;两轮叠加观察可见:第一轮分布集中于中等活性、细胞转化有限,第二轮则显著拓宽、深入低纳摩尔区间,且生化—细胞活性整体相关性良好。两位主角由此诞生:
| NCATS-SM0707(化合物 13) | 1.12 nM | 13.62 nM | ||||
| NCATS-SM0708(化合物 14) | 31.63 nM | 3.84 nM | ||||
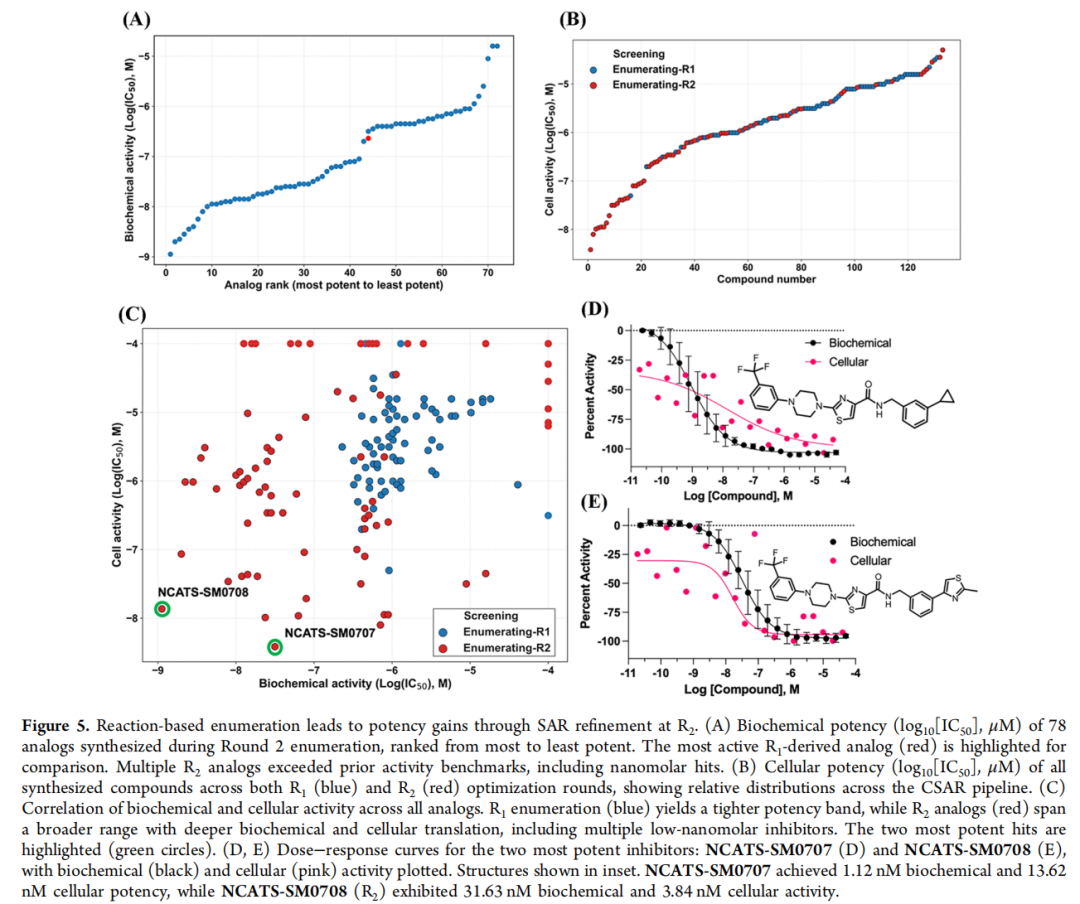
4.4 同工酶选择性谱
作者将两个先导物置于一整排近亲同工酶面前测试(ALDH1A1、1A2、1A3、ALDH2,以及同源约 70% 的 ALDH3A2)。按"家族内 > 30 倍即为优异"的标准,二者均达标:
| ALDH3A1 | 1.12 nM | 31.63 nM | |
| 相对 1A3 选择性 | > 2,816 倍 | > 25 倍 |
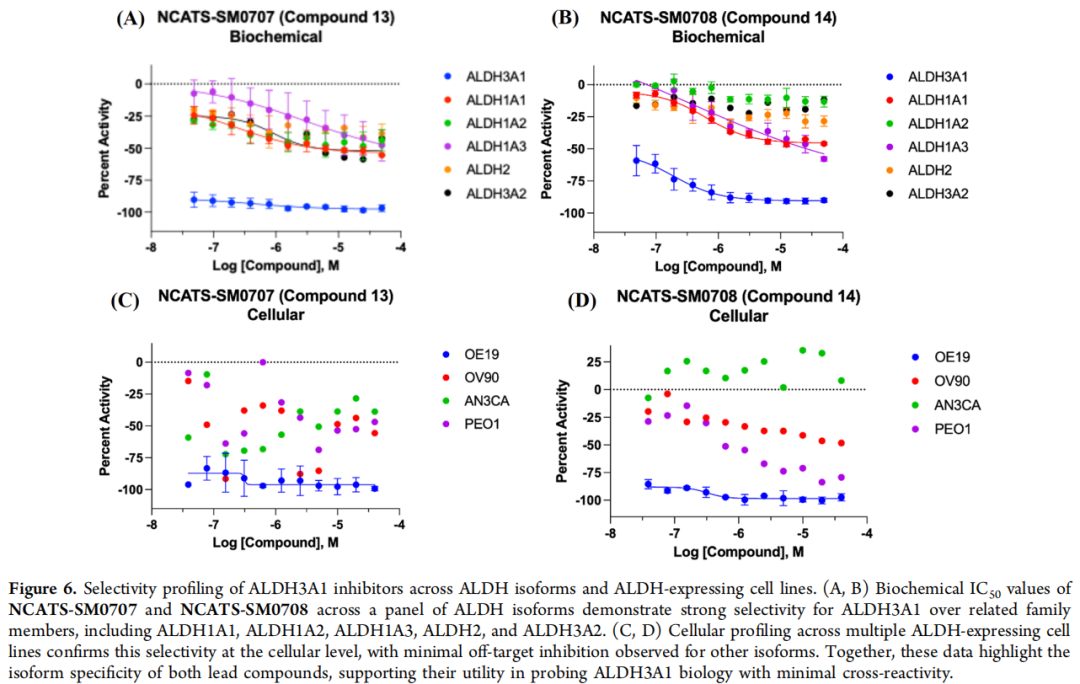
细胞层面亦印证:SM0707 在 OV90(1A1)、AN3CA(1A2)、PEO1(1A3)中弱/无活性;SM0708 在 AN3CA 中无活性,对 OV90、PEO1 仅中等(相对 OE19 的比值分别为 146、232)。效价低于 40% 或曲线质量差时不报告 IC₅₀。
4.5 抑制机制与结合验证
底物竞争实验:标准格式下苯甲醛设为约 1× K_m(200 μM)、NAD(P)⁺ 设为约 4× K_m(1000 μM,以偏离辅因子口袋)。将苯甲醛提高至约 20× K_m(4000 μM)后,两化合物剂量–反应曲线均明显右移,提示底物竞争性抑制;但因实验未配置评估辅因子竞争,是否存在混合模式仍待更多动力学研究。
光亲和标记 + 质谱:作者合成含双吖丙啶(diazirine) 的探针化合物 21(活性保留:生化 0.738 μM、细胞 15 nM),与重组 ALDH3A1 预孵后紫外交联,经 HPLC–MS/MS 蛋白质组学分析,检测到 +554.19 Da 质量位移,定位到肽段 WNAYYEEVVYVLE——该段为衬于活性位点的 α-螺旋上的修饰残基簇,投影到共晶结构后位于参考配体附近,从而正交支持化合物 13 结合于 ALDH3A1 经典底物口袋。
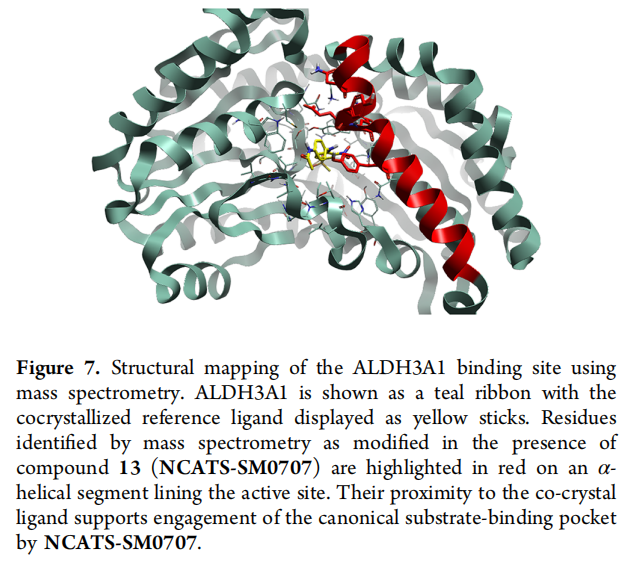
对接对照:比较原始苗头与 SM0707 的对接姿态显示,优化分子仍兼容同一结合区并保留噻唑-酰胺核心的放置;但 SM0707 给出两种均合理的取向(一种与苗头一致、一种翻转),提示精确几何不能仅凭对接确定,且未识别出单一足以解释全部活性提升的新增相互作用,更倾向于"整体口袋互补性改善"。能量层面的机制剖析需更高分辨率的结构/生物物理研究。
4.6 成药性与配体效率(诚实的一面)
多数强效化合物动力学水溶性偏低(pION μSOL,pH 7.4,普遍 < 1 μg/mL;吡啶基化合物 11 为 21.92 μg/mL,是少数例外),SM0708 的大鼠肝微粒体稳定性也仅中等。配体效率分析提供了重要补充:SM0707 相对苗头同时改善了配体效率(LE)与亲脂配体效率(LLE),说明其活性提升并非靠单纯增大分子体积或亲脂性而获得;SM0708 则保持强活性但效率指标更低,体现了该分支中"活性—物化代价"的不同平衡。
五、讨论与批判性评价
5.1 方法学层面的真正贡献
把这套工作放在方法学坐标系里看,其价值不止于几个 ALDH3A1 强效分子,而在于一个可迁移、可扩展、可复现的范式:
1. 缝合"直觉"与"规模" —— 不是用 AI 取代化学家,而是用反应枚举把化学家的逐位优化逻辑放大上千倍,候选名单最终仍由药化专家把关。 2. 可合成性是"内建"的 —— 90% / ~100% 的两轮合成成功率,反映枚举阶段就把反应可行性纳入考量,这恰是许多 in silico 流程被忽视的痛点。 3. 预测模型前置带来速度 —— 单轮即拿到 sub-230 nM,凸显在选型早期引入 QSAR 的实际收益。 4. 模块化与开放 —— DLCA 脚本、RF 对照、KNIME 枚举流程均开源于 GitHub,qHTS 数据公开于 PubChem,具备可复现性。
5.2 选择性从何而来
家族内选择性的获得有清晰的机制解释:整套药效团假设与对接过滤都构建在 ALDH3A1 口袋之上,奖励与其口袋体积、氢键拓扑(这些特征在家族内并不保守)互补的分子,从而把优化方向"拽"向适配 3A1、远离其它同工酶偏好的基序。换言之,"以 3A1 为中心的分流"本身就是选择性的来源——这是一个值得借鉴的设计思路。
5.3 局限与边界(应客观看待)
• 成药性短板:溶解度普遍偏低、部分化合物代谢稳定性中等,作者明确将两个先导物定位为"强效且选择性优异的先导物",而非可直接开发的候选药。 • 结合几何不确定:对接给出两种合理姿态,需更高分辨率结构或生物物理手段定论。 • 平台的依赖性:枚举库质量取决于商业砌块的多样性与反应模板可行性;QSAR 预测力取决于训练数据的量与质。 • 方法的局部性:本文仅演示两个反应/两个位点,尚未涉及 linker 替换、骨架跃迁等更广义的药化修饰(虽然作者强调框架可扩展)。
5.4 未来方向
作者提出的演进路径包括:将更先进的生成式模型用于骨架跃迁并与 SAR 扩展并行;把 ADMET 预测更早纳入优先级排序,实现真正的多参数优化(MPO);以及通过追加反应方案、在更多分子位点上枚举,探索"近乎无限"的化学空间。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢