

2026年6月11日,中国科学院上海药物研究所郑明月、张素林团队在 Nature Machine Intelligence 在线发表题为“Bridging three-dimensional molecular structures and artificial intelligence with a conformation description language”的研究论文。该研究提出分子构象描述语言 ConfSeq,将三维分子构象编码为离散标记序列,从而把构象预测、从头三维分子生成、形状条件分子生成和三维分子表征学习等任务统一转化为序列建模问题,并在多个基准测试和药物发现案例中系统评估了其性能与应用价值。

背景
人工智能(artificial intelligence, AI)正在改变多个学科领域的研究方式。语言模型(language models, LMs)通过自监督学习从大规模标记序列中捕捉复杂模式,并在通用人工智能系统(如 GPT-4 和 Gemini)以及领域专用模型(如 ESM、GeneFormer、NT、xTrimoPGLM 和 Evo)中得到广泛应用,为科学研究和产业应用提供了新的计算工具。
在药物和化学研究中,化学语言模型(chemical language models, CLMs)已成为重要研究方向。通过将分子拓扑结构编码为离散标记序列,例如 SMILES 和 SELFIES,CLMs 能够支持分子翻译、从头生成和表征学习等二维分子建模任务,并在多项基准测试中取得具有竞争力的表现(图1a)。基于这一范式,研究团队近期还开发了反应描述语言 ReactSeq,用于将化学转化过程编码为显式标记序列。
分子性质不仅取决于二维拓扑连接关系,也受到三维空间构象的显著影响;二者共同决定分子的理化性质、反应活性及其与生物靶标的相互作用。因此,面向三维分子结构的有效表示和生成方法,是 AI 驱动化学与生命科学研究中的关键问题。与二维分子建模相比,语言模型在三维场景中的应用仍受到表示方式限制。目前,该领域主要由图扩散模型等几何生成方法推动,但这类方法通常需要专门的空间结构设计,并可能面临生成效率、结果筛选和内置评分机制等方面的限制。由此,如何将化学语言模型范式从二维分子结构自然拓展到三维分子构象,成为值得探索的问题。
已有研究对这一方向进行了初步尝试。例如,Lingo3DMol 利用 LMs 生成片段 SMILES,并结合辅助空间模块预测片段位置以构建三维分子。该设计具有一定效果,但依赖与标准语言模型不完全兼容的专用空间组件,限制了方法的通用性;同时,片段级构建方式可能忽略环系构象柔性,从而影响三维建模精度。近期也有研究尝试采用纯语言模型方法,将原子坐标编码为字符串并与 SMILES 拼接以生成三维分子。这类方法在概念上直接,但笛卡尔坐标表示容易受到平移和旋转影响,缺乏 SE(3) 不变性,也缺少系统的标记化策略;此外,语言模型并不天然擅长处理连续数值数据,直接使用坐标字符串可能影响空间关系学习。相关方法也仍需要在更多样化的数据集和任务上进行系统评估。
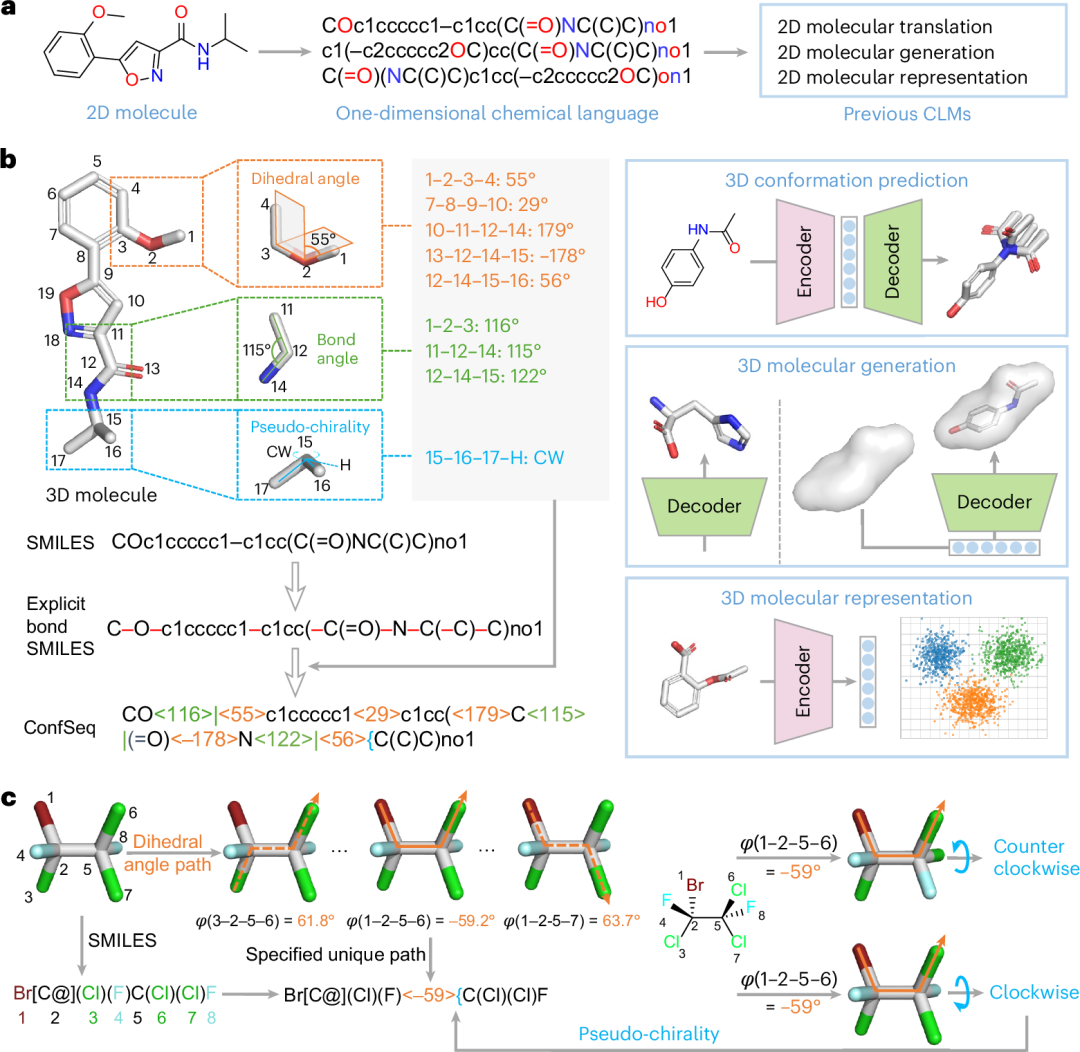
将 LMs 扩展至三维分子建模所面临的核心挑战,是缺乏一种高效、稳定且适合标记化学习的分子构象表示语言。理想的表示方式需要把连续、无序的几何信息转化为离散序列标记,同时尽量保持 SE(3) 不变性、简洁性和可解释性。为解决这一问题,研究团队开发了 ConfSeq。该构象描述语言利用三类关键内在几何元素编码分子构象:二面角、键角和伪手性描述符。这些几何元素被离散化为标记,并策略性地整合到 SMILES 框架中。该设计既保留了 SMILES 的可读性,也支持基于 SMILES 枚举的数据增强,并通过把几何标记与原子、化学键上下文关联起来,帮助 LMs 学习几何—结构关系(图1b)。
借助 ConfSeq,本研究将构象预测、三维分子生成和三维分子表征等多类三维分子建模任务重构为序列建模问题。基于 ConfSeq,研究团队使用标准 Transformer 架构在多项任务中进行了基准评估,并观察到优于既有方法的表现。与此同时,这一序列化范式可利用语言模型在生成效率和自回归概率评分方面的特点,为候选构象或生成分子提供内在置信度参考。研究还通过虚拟筛选和体外实验验证了 ConfSeq 的实用性,发现了多个新型 STING 抑制剂和 ALDH1B1 抑制剂,IC50 值范围为 0.338-3.51 μM。总体而言,这些结果表明,ConfSeq 为语言模型处理三维分子建模任务提供了一种可行的表示框架。
结果与讨论
ConfSeq概述
ConfSeq 通过为三类关键几何参数引入自定义标记,对分子构象进行系统编码(图1b)。
二面角由四个连续成键原子(i-j-k-l)定义,用于量化原子 i-j-k 和 j-k-l 所形成平面之间的夹角。对于给定中心键,可能存在多条二面角路径和多个角度值,从而产生编码歧义(图1c)。为避免这一问题,研究团队设计了确定性的路径选择算法,为每个可旋转键分配唯一的二面角路径。所得角度值被四舍五入为最接近的整数,并以尖括号封装(例如“<70>”),随后插入 SMILES 中对应键标记的位置,从而实现二面角信息的唯一且确定性编码。
键角由三个连续成键原子(i-j-k)定义,用于测量中心原子 j 处键 j-i 与 j-k 之间的夹角。与二面角类似,键角值(0°-180°)被四舍五入为整数,以尖括号封装,并添加后缀“|”以示区分。该标记在 ConfSeq 序列中紧跟中心原子标记之后插入。如图1b所示,“<115>|”标记位于原子12(“C”)的标记之后,从而建立直接的几何—原子上下文。
伪手性用于补充仅靠二面角难以描述的非手性原子周围构象歧义。例如,在图1c中,二面角 1-2-5-6 定义了原子1至6的取向,但不能确定原子5周围取代基7和8的排列方式,因此仍存在两种可区分构象。伪手性定义为从最低编号取代基方向观察时,非手性原子周围取代基的顺时针或逆时针排列。在 ConfSeq 中,这两种状态分别以“{”和“}”表示,并根据原子编号置于对应二面角标记之后(例如“<59>{”)或之前(例如“{<59>”)。该描述符与二面角结合后,可唯一指定可旋转键周围取代基的空间排列。
与基于绝对坐标的既有三维分子语言相比,ConfSeq 以内坐标表示分子,有助于捕获有效构象所在的低维流形,并降低建模复杂度。通过将内坐标标记嵌入 SMILES 框架,ConfSeq 可以同时保留化学拓扑上下文和三维几何信息。相较于仅考虑可旋转键二面角的图式内坐标模型,ConfSeq 进一步纳入环内二面角、键角和伪手性,将平均重构误差从 0.65 Å 降至 0.23 Å。
基于ConfSeq的分子三维构象预测
为展示 ConfSeq 的适用性,研究团队首先将其用于分子构象预测。研究采用标准 Transformer 架构,将分子 SMILES 翻译为 ConfSeq 表示,并可由该表示确定性解码得到三维分子构象。模型在 GEOM-Drugs 数据集上训练和评估,并采用 Coverage(COV)和 Matching(MAT)评分量化精确率(P)和召回率(R)。
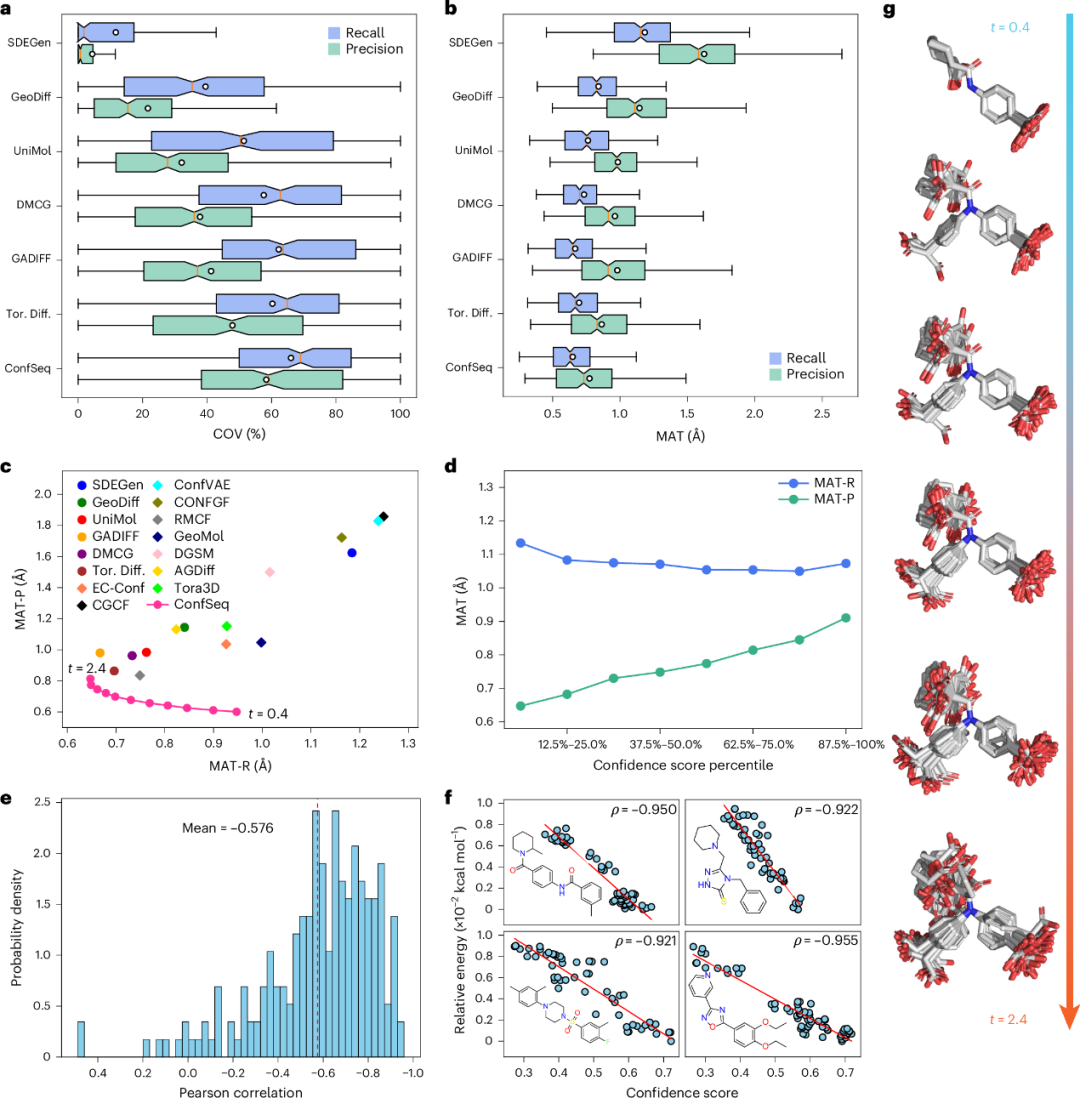
如图2a-b所示,ConfSeq 模型在各项评价指标上均表现良好。与既往表现最优的方法 Tor. Diff. 相比,在 0.75 Å 阈值下,COV-P 从 47.9% 提高至 58.4%,MAT-P 从 0.86 Å 降至 0.77 Å。此外,借助语言模型的采样机制,ConfSeq 能够通过调整采样温度来调节召回率—精确率权衡(图2c)。在不同温度下,ConfSeq 相比基准方法形成了更优的 MAT-P/MAT-R 帕累托前沿(Pareto front)。图2g中的代表性构象进一步显示了采样温度对结构多样性的调节作用。这些结果也提示,构象预测任务中的模型性能更适合通过完整的召回率—精确率权衡曲线进行评价,而不宜仅依赖单点指标。
ConfSeq 的另一项特点是能够根据自回归逐步概率为预测构象分配定量置信度评分。随着置信度评分提高,生成构象的 MAT-R 保持稳定,而 MAT-P 明显降低(图2d),说明模型评分可用于优先筛选更高质量的候选构象。为评估模型评分是否反映能量合理性,研究团队进一步在测试集中的量子化学优化构象上进行分析。对于多数分子,置信度评分与分子能量呈负相关,平均 Pearson 相关系数为 -0.576;对于可旋转键较少的分子,这一关系更强,在部分情况下相关系数低于 -0.9(图2e-f)。研究人员还在规模更大、化学多样性更高的 QMugs 数据集上进行测试,结果显示,基于 ConfSeq 的模型仍优于此前表现较好的方法,说明其在多样化化学空间中具有一定稳健性。
基于ConfSeq的无条件三维分子生成
在验证 ConfSeq 用于分子构象预测的可靠性后,研究团队进一步将其扩展到无条件三维分子生成任务。该任务是从头药物设计中的基础问题之一,目标是在无外部输入约束的条件下,从已学习的数据分布中生成三维分子结构,从而探索化学空间。研究团队采用仅解码器(decoder-only)Transformer 架构,并主要与代表性的图扩散方法在二维和三维评价指标上进行基准比较。
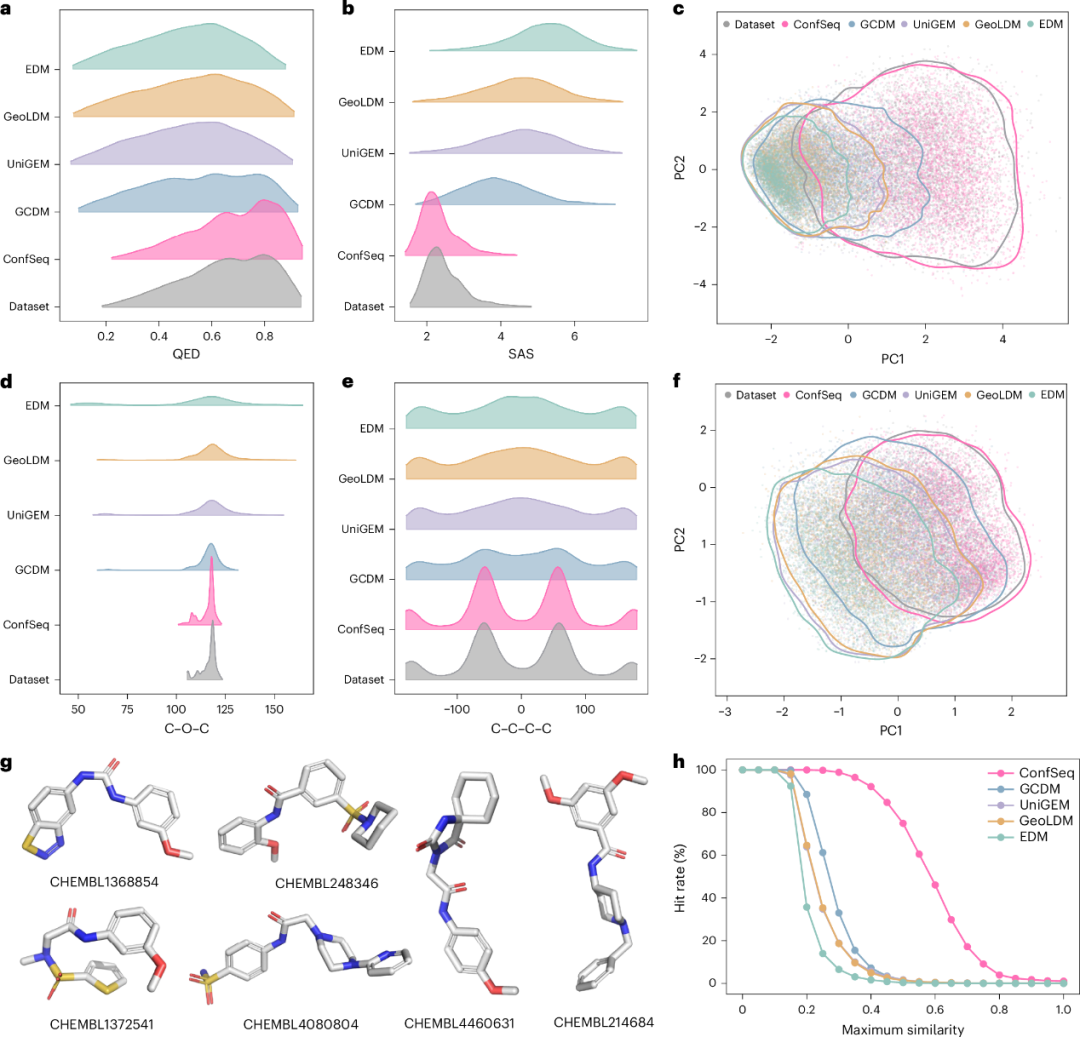
研究团队首先从二维层面评估模型性能。ConfSeq 实现了接近 100% 的化学有效性(validity)和较高唯一性(uniqueness),优于其他基线模型。此外,部分基线模型在定量类药性估计(QED)和合成可及性评分(SAS)等类药性质上与训练集分布存在偏离,而 ConfSeq 能够更接近地复现这些性质分布(图3a-b)。ChemNet 嵌入分析进一步显示,ConfSeq 生成分子与训练集分子更为相似,提示模型较好地捕获了训练数据中的常见结构基序(图3c)。
在三维评价中,ConfSeq 生成的三维分子结构不仅在物理上更合理,而且与训练数据的构象分布更接近。ConfSeq 达到 82.3% 的 PoseBusters 有效性(PB-validity),较此前 SOTA 方法提高 6%,并将最小 RMSD 降至 0.1024,降低幅度为 35%。与此前 SOTA 方法相比,ConfSeq 将二面角 MMD 从 0.0304 降至 0.0070,将键角 MMD 从 0.1048 降至 0.0543。如图3d-e所示,基线模型生成的角度分布往往被过度平滑化,并主要呈现单峰特征;相比之下,ConfSeq 能够更准确地捕捉训练数据中的多峰分布特征。基于 E3FP 的分析进一步显示,在所比较的方法中,ConfSeq 生成分子与训练分布的总体相似性最高(图3f)。值得注意的是,得益于语言模型的推理效率,研究团队的方法在 GPU 上的总体采样速度约为当前 SOTA 扩散模型(GCDM)的 285 倍。
最后,为评价各模型发现潜在生物活性分子的能力,研究团队计算了生成的新分子与 ChEMBL 数据库条目的最大相似性。如图3g所示,在 10,000 个生成样本中,ConfSeq 能够命中 91 个已知活性分子,而其他模型最多命中 13 个活性分子,且主要为原子数较少的简单结构。综合这些结果,ConfSeq 能够生成具有较好二维/三维有效性的分子,并在理化性质和构象特征上与训练集分布保持较高一致性。
基于ConfSeq的形状条件三维分子生成
鉴于 ConfSeq 在无条件三维分子生成中表现良好,研究团队进一步将其扩展到形状条件生成任务。该任务更具挑战性,要求生成分子在满足预设三维形状约束的同时保持结构新颖性,因此与骨架跃迁和专利空间探索等药物设计场景密切相关。本研究将分子表面表示为点云,并利用旋转不变表面卷积(Rotation-Invariant Surface Convolution, RISConv)提取几何不变表面特征。随后,这些特征由 Transformer 解码器处理,生成可解码为三维分子的 ConfSeq 序列(图4a)。
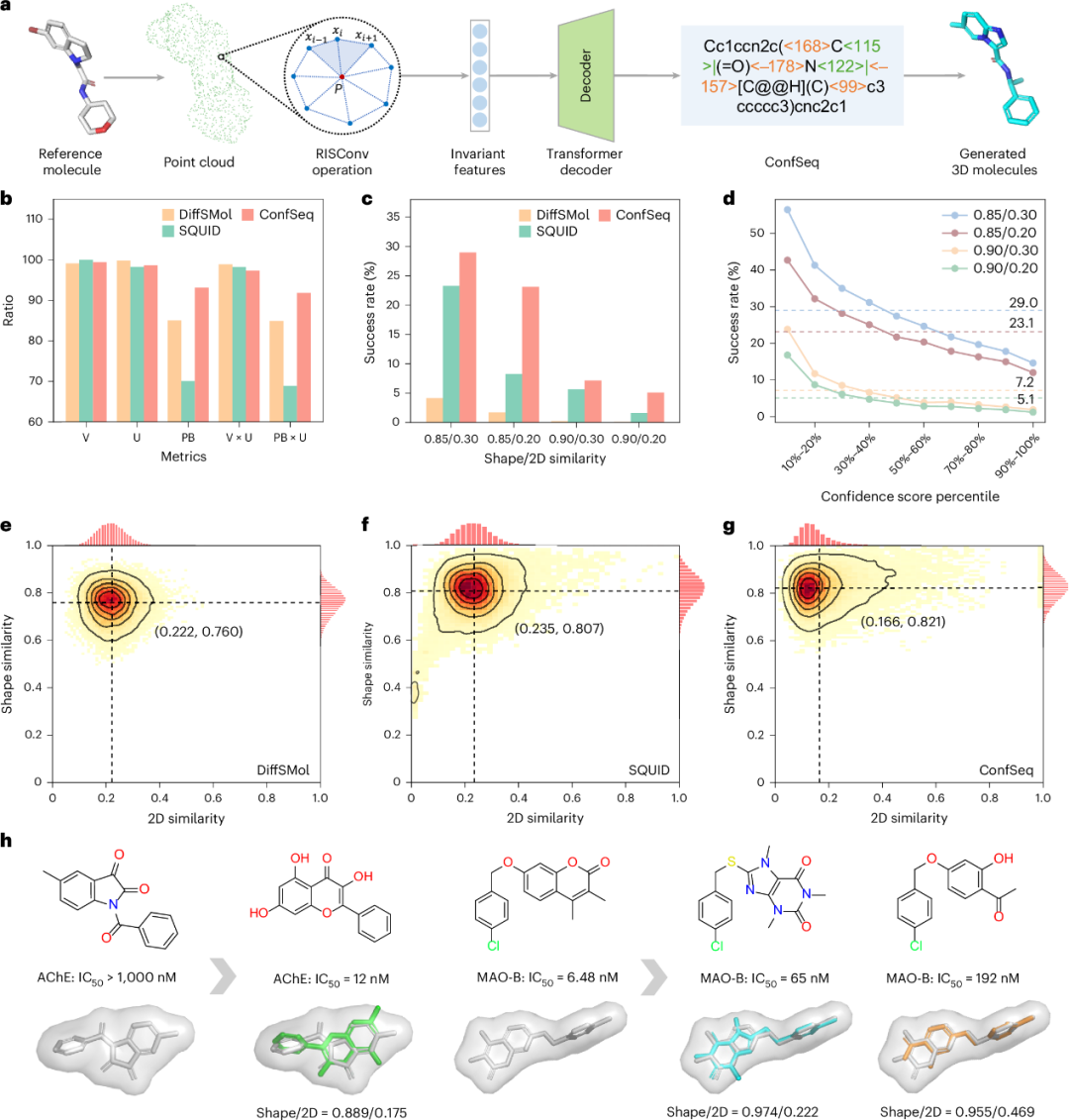
与无条件生成任务一致,研究团队从二维和三维两个层面评价模型性能。ConfSeq 达到 93.1% 的 PB-validity,高于 DiffSMol(85.1%)和 SQUID(70.1%),并保持接近 100% 的化学有效性(图4b)。研究团队进一步从三维形状和二维结构两个角度量化生成分子与参考分子的相似性。如图4e-g所示,ConfSeq 生成分子表现出更高的形状相似性和更低的二维相似性,说明其在几何约束下更有利于获得骨架新颖的候选分子。在同时要求高形状相似性和低二维相似性的组合标准下(形状相似性 >0.85 或 >0.9;二维相似性 <0.3 或 <0.2),ConfSeq 的成功率分别为 5.1%、7.2%、23.1% 和 29.0%,较此前 SOTA 方法提高 24%-210%(图4c)。
进一步地,模型同样展现出较强的打分能力,按置信度评分筛选候选分子可提升性能;在最严格标准(形状相似性 >0.9 且二维相似性 <0.2)下,选择前 10% 候选分子可将命中率从 5.1% 提高至 16.8%(图4d)。同时,基于 ChEMBL 活性分子数据库的回顾性案例显示,ConfSeq 能够生成形状和生物活性相似、但骨架结构不同的分子(图4h)。总体而言,这些结果支持 ConfSeq 作为形状引导分子生成框架的有效性,并显示其在骨架跃迁任务中的应用潜力。
基于ConfSeq的三维分子表征学习
除三维分子生成外,研究团队进一步将 ConfSeq 扩展至三维表征学习。研究采用仅编码器(encoder-only)Transformer 从 ConfSeq 序列中获得固定维度嵌入。这些嵌入通过度量学习框架训练,使向量间距离能够反映分子三维相似性。模型在来自 ChEMBL 和 BindingDB 的 280 万个分子上训练,其构象由 RDKit 生成。
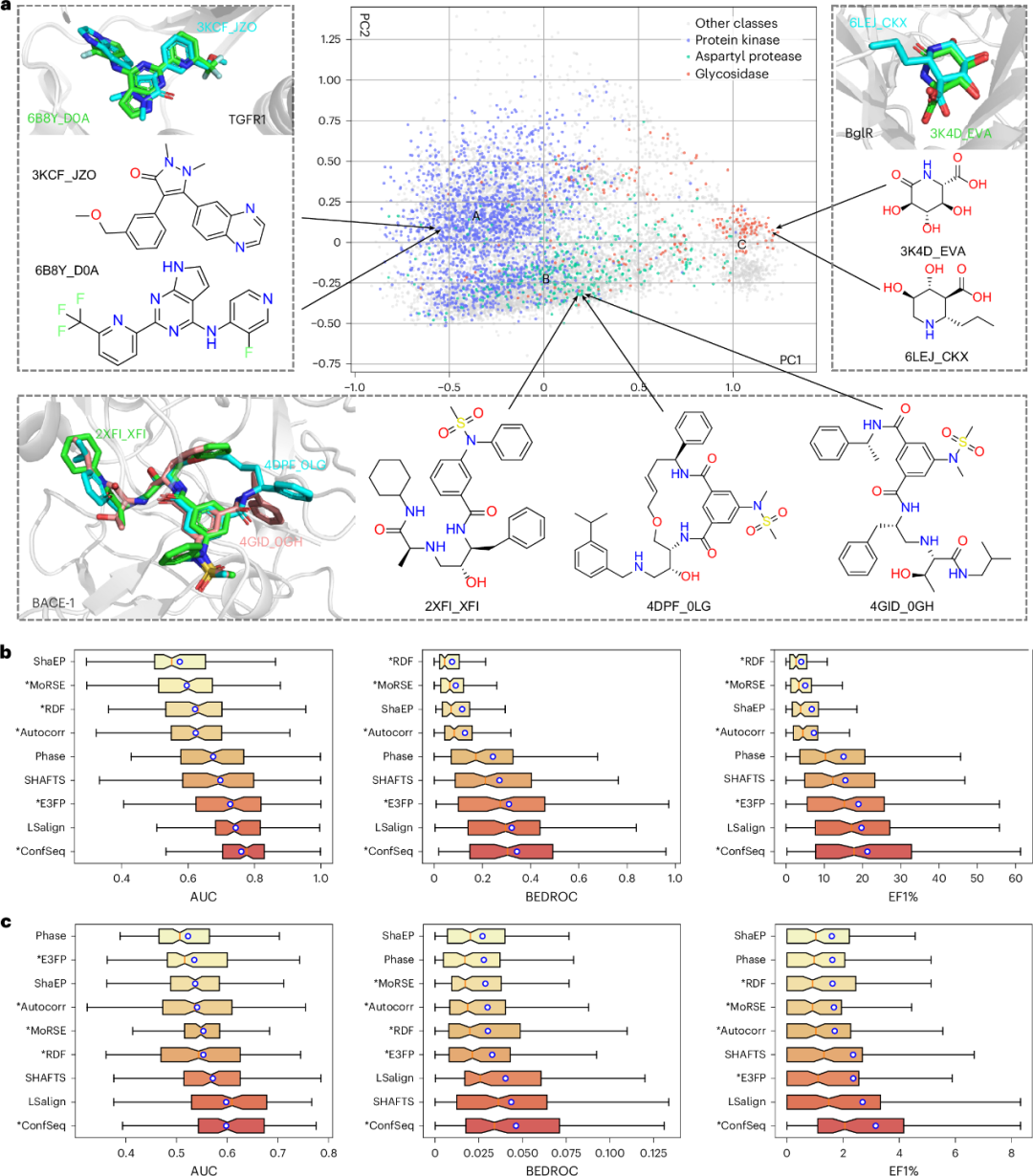
研究团队利用训练好的模型从 PDB 数据库推理得到配体表征,并通过 PCA 进行可视化。如图5a所示,基于 ConfSeq 的表征能够将结合至同一蛋白口袋的分子聚类,即使这些分子的二维结构存在明显差异。例如,模型捕获了两个线性 BACE-1 抑制剂(2XFI_XFI 和 4GID_0GH)与一个结构不同的大环抑制剂(4DPF_0LG)在结合构象上的三维相似性,表明模型学习到的表征能够编码超越二维拓扑的构象相似性。PDB 中的配体还进一步聚成三个富集特定靶标类别的簇:激酶抑制剂(簇A)、天冬氨酸蛋白酶抑制剂(簇B)和糖苷酶抑制剂(簇C),每簇均呈现与其已知配体类别一致的特征结构。这些结果共同表明,基于 ConfSeq 的表征能够捕获靶向同一蛋白家族配体之间保守的三维结构模式,而这些模式仅依赖二维分子表征通常难以辨识。
除定性可视化外,研究团队还在 DUD-E 和 PCBA 数据集上定量评估了基于 ConfSeq 的表征在基于配体虚拟筛选中的效用。如图5b-c所示,ConfSeq 在 DUD-E 上取得 AUC 0.76、BEDROC 0.34、1.0% 富集因子(EF1%)21.25 的结果;在 PCBA 上这些值分别为 0.60、0.046 和 3.16,优于三维分子指纹方法(E3FP 和 MORSE)以及基于比对的三维相似性工具(LSalign 和 SHAFTS)。除筛选性能外,ConfSeq 还通过在表征空间中直接测量相似性,替代计算开销较大的成对叠合,因此相较基于比对的方法具有明显效率优势。
为支持大规模应用,研究团队利用模型预先计算了 ZINC In-Stock 和 PubChem 数据库中三维化合物的嵌入。基于这些表征,在单个 CPU 核心上即可在约 3 分钟内计算一个查询分子与 9800 万个 PubChem 化合物之间的三维相似性,从而支持大规模虚拟筛选。综上,这些结果表明,ConfSeq 是一个可扩展的三维分子表征框架,能够捕获具有生物学意义的构象相似性,并支持此前较难实现规模的高效虚拟筛选。
ConfSeq促进新型STING和ALDH1B1抑制剂的发现
为在真实药物发现场景中评估 ConfSeq,研究团队首先针对 STING(stimulator of interferon genes,干扰素基因刺激因子)开展基于配体的虚拟筛选。STING 是重要治疗靶标,其构象柔性和二聚体界面结合模式使基于结构的筛选具有挑战性。研究团队以 14 个结构多样的已知 STING 调节剂为查询分子,利用基于 ConfSeq 的三维表征筛选 ChemDiv 化合物库,按三维相似性排序,对前 20,000 个候选分子进行聚类以去除冗余,并经可获得性筛选后选择 92 个化合物进行实验验证(图6a)。
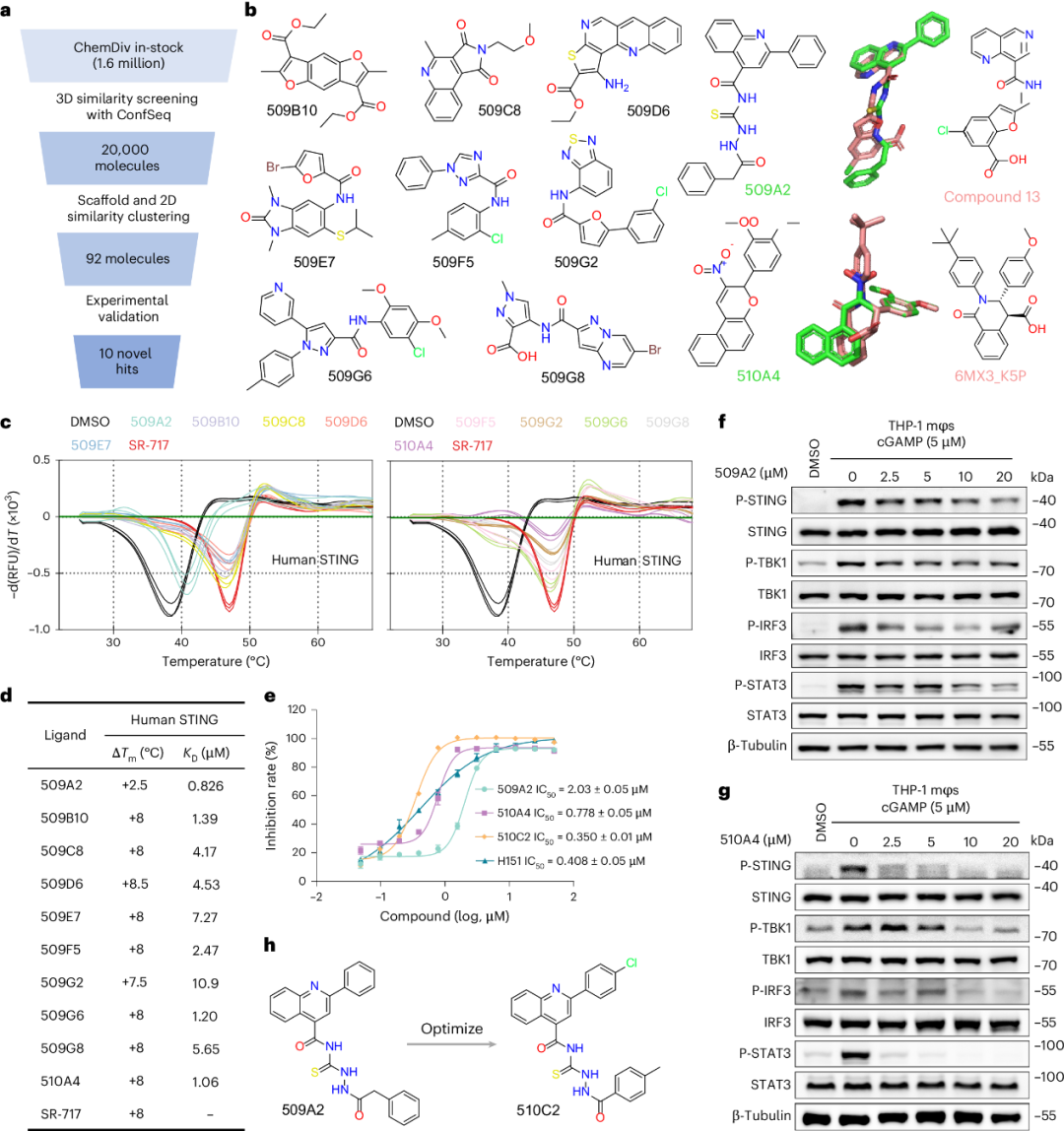
研究团队利用蛋白热迁移(protein thermal shift, PTS)和表面等离子体共振(surface plasmon resonance, SPR)实验进行生物物理表征,确认 10 个化合物可与 STING 结合,总体命中率约为 11%(图6b)。这些命中化合物的热稳定性迁移(ΔTm)范围为 +2.5 °C 至 +8.5 °C,与人源 STING 相互作用的解离常数(KD)为 0.826-10.9 μM(图6c-d)。其中,亲和力最高的两个候选分子 509A2 和 510A4 对 cGAMP 诱导的 STING 激活表现出拮抗活性,半数最大抑制浓度(IC50)分别为 2.03 μM 和 0.778 μM(图6e)。两个化合物还以剂量依赖方式抑制多种 STING 激动剂(cGAMP、diABZI 和 SR-717)诱导的通路激活,表现为 STING、TBK1、IRF3 和 STAT3 磷酸化水平降低(图6f-g)。细胞毒性分析进一步给出生长抑制浓度(GI50)和安全指数(SI = GI50/IC50),其中 509A2 的 SI > 25,510A4 的 SI 约为 4。总体而言,这些结果验证了所鉴定 STING 抑制剂的靶向抑制活性和进一步优化价值。
值得注意的是,509A2 和 510A4 与参考化合物(Compound 13 和 6MX3_K5P)具有较高的三维构象相似性,但二维分子骨架明显不同,提示 ConfSeq 筛选结果实现了骨架跃迁(图6b)。这扩展了 STING 调节剂的化学多样性,也为规避既有结构空间提供了候选思路。随后,研究团队对 509A2 的商业可得类似物进行了实验评估,其中类似物 510C2 的活性提高约 6 倍(IC50 = 0.350 μM),且细胞毒性较低(图6e, 6h)。其抑制活性和毒性特征优于阳性对照 H151,显示出作为后续临床前研究先导化合物的潜力。
为进一步评估 ConfSeq 的实际应用价值,研究团队还针对 ALDH1B1 开展了另一轮基于配体的虚拟筛选,并发现 3 个经实验验证的抑制剂,其 IC50 值范围为 0.338-3.51 μM。以上两个案例共同说明,ConfSeq 在基于配体的虚拟筛选中具有可迁移的应用潜力,并可用于不同治疗靶标的候选分子发现。
结论
本研究围绕将 CLMs 用于三维分子建模所面临的关键挑战展开,即缺乏针对三维分子结构的稳健一维表示。为此,研究团队开发了 ConfSeq。作为一种分子构象描述语言,ConfSeq 将 SMILES 与三维分子关键内在几何信息(二面角、键角和伪手性)融合,在保持 SE(3) 不变性的同时,保留了 SMILES 的可读性和简洁性。
通过将构象预测、无条件和形状条件生成以及三维表征学习等多种三维分子建模任务重新定义为序列建模问题,ConfSeq 使标准 Transformer 架构能够在基准数据集上取得具有竞争力的表现,并在多个任务中优于既有方法。与当前主流图扩散方法相比,ConfSeq 利用了语言模型在推理效率和自回归评分方面的特点,既可提高生成效率,也可为候选结构筛选提供内在评分。更重要的是,ConfSeq 的实用价值通过体外实验得到验证,研究团队发现了多个活性良好的新型 STING 和 ALDH1B1 抑制剂。
总体而言,本研究为“语言模型如何处理三维分子任务”提供了一种新的技术路径。ConfSeq 在语言模型和三维分子结构之间建立了可操作的表示框架,有望为 AI 驱动的分子建模、药物发现和分子设计提供新的工具选择。

内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢