

论文全称:Site4Drug: Predicting Drug-Binding Target Sites with an AI Agent
发表会议:ICML 2026 Workshop on Generative and Agentic AI for Biology(GenBio)
作者:Taehan Kim†(UC Berkeley)、Sarrah Rose Mikhail Leung、Bharat Mekala、Jeongbin Park†(University of Michigan)
代码:https://github.com/winterrykim/Site4Drug_Demo
arXiv:2506.01816
一、背景与动机:一个被忽视的上游瓶颈
1.1 现有流程的缺口
现代计算药物发现流程已高度工具化。从分子对接(docking)、虚拟筛选,到近年兴起的深度学习生成模型,绝大多数方法都有一个隐含前提:结合位点已知。
以下两类代表性工具可以说明这一点:
• BoltzGen(Stark et al., 2025):基于扩散模型的结合子设计工具,需要用户预先给定靶点区域; • BindCLIP / DrugCLIP(Qiao et al., 2026;Gao et al., 2023):对比学习式口袋-分子打分模型,同样以预定义位点为输入。
当下游筛选失败时,团队往往无从判断:究竟是结合模型失效,还是位点选错了?这一责任归因困难部分源于"位点选择步骤几乎从不被记录,或完全依赖经验直觉"。
1.2 膜蛋白的特殊困难
膜蛋白是一大类高价值药靶(约 60% 的上市药物靶向膜蛋白),但它们的成药区域分析尤为复杂:
| 拓扑歧义 | |
| PTM 遮蔽 | |
| 可及性约束 | |
| 结构稀缺 |
1.3 现有工具的局限
fpocket(Le Guilloux et al., 2009)和 RAPID-Net(Balytskyi et al., 2025)是小分子口袋发现领域的代表工具,前者基于 Voronoi 球填充识别几何空穴,后者用学习的结构表示预测结合位点。两者的共同局限在于:
1. 均以三维结构为必须输入,新靶标往往无结构可用; 2. 只能发现"经典"结合口袋,无法支持抗体表位等替代模态; 3. 不能整合 PTM、保守基序、二硫键等元数据约束; 4. 输出不可解释,失败难以归因。
IEDB 等免疫表位数据库可提供参考,但其收录的表位来自高度专门化的免疫实验(T/B 细胞测定),并非系统枚举全部成药位点,也不是治疗性位点选择的金标准。
论文的核心主张:将靶点区域选择重新定义为一个约束优先、证据整合的决策问题,而非直觉驱动的操作,并通过 LLM 智能体实现推理过程的可审计与可调试。
二、方法:Site4Drug 的架构设计
2.1 问题形式化
输入:一条长度为 的氨基酸序列 (无需三维结构)。
输出:结构化的位点选择报告,包含:
• 推荐结合模态:,附带置信度与不确定性说明; • epitope:抗体/肽类结合子附着位点,通常位于膜蛋白胞外突出区域;• pocket:小分子结合口袋,通常在胞内蛋白或膜通道中;• other:证据指向非标准或混合模式(如可及性模糊、约束竞争);• 排名候选区域列表 ,每项包含: • 可及性/拓扑标签( tmd/restricted或outside/exposed);• 证据摘要(疏水性、跨膜重叠、PTM 掩码、基序命中、半胱氨酸/二硫键上下文); • 风险标记( TM-overlap、PTM-overlap、glyco-mask-overlap、disulfide-constrained、motif-overlap等);• 完整审计日志:记录每一步推理所依据的证据与约束。
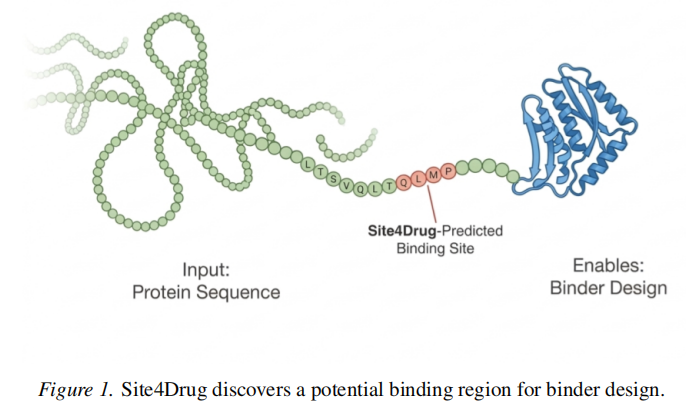
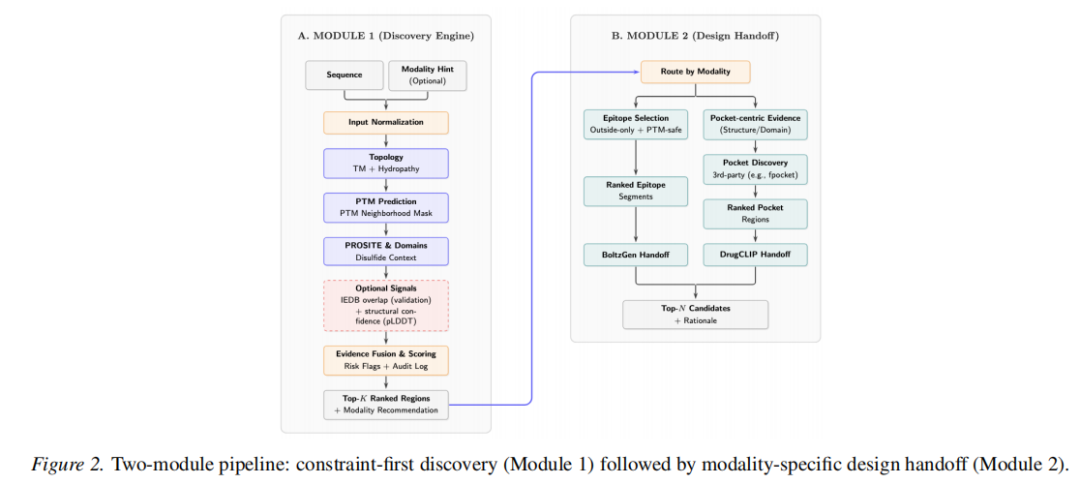
2.2 Module 1:证据聚合与区域发现
整个发现引擎的核心在于从序列中提取三类可行性感知信号,构建残基级和区域级证据摘要。
信号 I:粗粒度拓扑 + 疏水性
Site4Drug 使用经典的 Kyte–Doolittle 滑动窗口疏水性剖面(窗口大小 = 19)结合启发式跨膜区域检测器(TM detector),仅从序列推导粗粒度可及性先验:
其中 为 Kyte–Doolittle 疏水指数(如 Ile = 4.5,Arg = −4.5)。检测到的跨膜段被标注为 tmd/restricted,胞外/胞内非跨膜区域标注为 outside/exposed,置信度由疏水性均值与 TM 阈值的差距决定。
信号 II:PTM 预测 + 邻域掩码
PTM 位点通过 MusiteDeep(Wang et al., 2020)后端 + 规则增强获得,覆盖:磷酸化(Ser/Thr/Tyr)、N-连接糖基化、泛素化、N6-乙酰赖氨酸、羟脯氨酸等多种修饰类型。
每个预测的 PTM 位点被扩展为一个类型化局部掩码(例如,位点 211 处的磷酸化 → 掩码覆盖残基 208–214),并记录候选区域与该掩码的重叠情况(重叠计数、PTM 类型分布、局部 PTM 密度)。
PTM 掩码的设计逻辑在于:高度修饰的区域在折叠蛋白的生理状态下往往结构受约束或被屏蔽,是药物接触的不利区域。
信号 III:基序命中 + 半胱氨酸/二硫键代理
• ScanProsite(De Castro et al., 2006):检索 PROSITE 数据库,记录功能基序(如 EGF 结构域、N-糖基化位点)的命中区间;与已知基序重叠的候选区域标注 motif-overlap——这类区域往往在进化上高度保守,靶向存在潜在选择性风险;• 半胱氨酸计数:作为二硫键约束区域的轻量级代理指标;区域内 个半胱氨酸触发 disulfide-constrained标记(注意:当前版本不做精确二硫键配对预测,仅作为结构风险代理)。
候选生成与排名
以上三类信号的压缩摘要连同完整氨基酸序列一起送入 LLM,直接以 JSON 格式提议并排名候选区间。每个候选经过合法性过滤后,被系统化地标注拓扑证据、PTM 重叠、基序重叠、半胱氨酸数量、类型化风险标记和启发式得分。
概念上,排名逻辑遵循以下模态特定启发式:
其中:
• :基础偏好分(表位模式偏好极性、非跨膜、PTM 稀疏窗口;口袋模式更重视疏水性,PTM 惩罚较弱); • 、、:来自跨膜重叠、PTM 掩码重叠、基序上下文的约束感知惩罚项。
风险向量 则以类型化标记的形式输出,并列于最终报告中。完整的风险标记触发规则见下表:
TM-overlap | |
PTM-overlap | |
glyco-mask-overlap | |
PTM-dense | |
disulfide-constrained | |
hydrophobic-core | |
motif-overlap |
2.3 多智能体专家评审(Specialist Panel)
候选区域初步排名完成后,系统引入一个多智能体二次审裁层,由四个角色构成:
BioAgent → 生物可行性审查(拓扑合理性、PTM 约束解读)
ChemAgent → 化学合理性审查(疏水性、溶解度、成药性)
RiskAgent → 风险综合评估(安全性、保守性、可及性冲突)
DecisionAgent → 综合三方 claim→evidence→impact 批评,输出最终模态决策与调整后排名每个专家智能体输出标准化 JSON 格式的评审意见:
{
"agent": "ChemAgent",
"modality_votes": {"epitope": 0.2, "pocket": 0.7, "other": 0.1},
"candidate_adjustments": [
{"candidate_id": "c_0001", "delta": -0.15, "reason": "高疏水性可能降低表位可及性", "evidence": ["mean_hydropathy=0.97", "hydrophobic_fraction=0.60"]}
],
"risk_flags": ["hydrophobic-core"],
"summary": "..."
}DecisionAgent 在综合三方意见时严格限定在已有上下文证据范围内,避免引入新的外部假设,确保推理链条的可追溯性。
2.4 Module 2:模态特定的设计移交
Module 1 的排名输出可直接作为下游设计工具的接口:
表位模式(Epitope Mode)
→ 将 Top-K 表位区间及其拓扑/PTM 风险注释传入肽/抗体结合子生成工具(如 BoltzGen),进行肽段设计;
→ 生成的候选结合子可用 AlphaFold3 进行多聚体结构预测和 LIS(局部相互作用分数)评估。
口袋模式(Pocket Mode)
→ 从 AlphaFold3 结构中提取预测口袋区域的原子坐标,构建局部口袋;
→ 送入 DrugCLIP 进行基于对比学习的虚拟筛选;
→ 必要时使用 Boltz2 做配体-蛋白结合亲和力评估(AlphaFold3 当前不支持通用配体预测)。
此外,Site4Drug 的表位输出也已与 Diffuse Bio Sandbox 集成,支持纳米抗体(Nanobody)和单链可变区(scFv)的结构条件化设计。
三、实验评估:多模态验证框架
3.1 数据集构建
由于靶点位点发现领域缺乏广泛认可的基准测试集,研究者自行构建了一个覆盖三种模态的评估数据集:
| Group S | ||
| Group A | ||
| Group AS | ||
| 合计 |
验证指标:超几何分布检验(p < 0.05),评估预测区域与参考位点的残基重叠是否显著高于随机期望。提供 Top-1 和 Top-5 两个层级的结果。
3.2 小分子口袋评估(Group S + AS,n = 63)
结果概览:
| Site4Drug | 20/63(31.7%) | ||
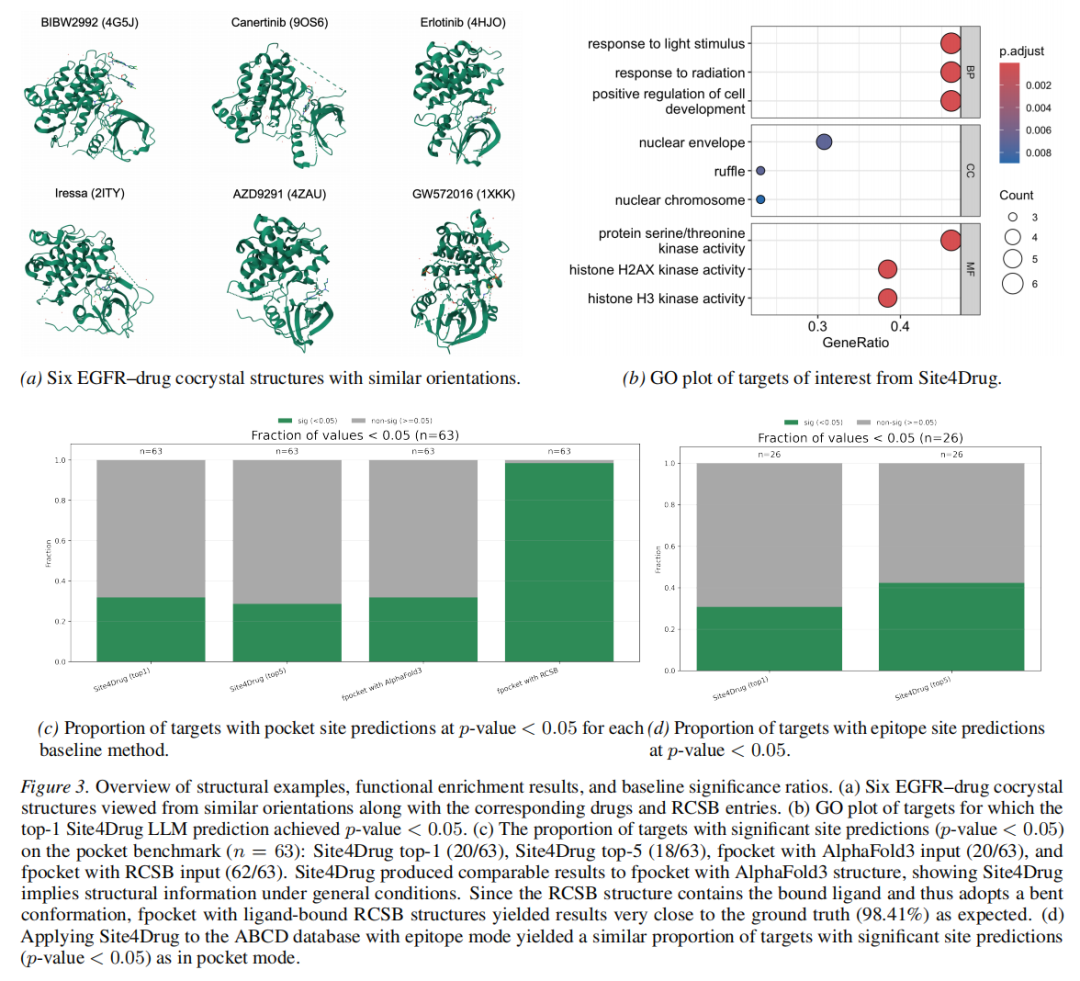
关键发现:
1. 与 fpocket(AlphaFold3)性能相当:Site4Drug 仅凭序列特征,在不使用任何蛋白质三维结构的前提下,达到了与需要 AF3 结构作为输入的 fpocket 相近的性能,体现了序列级证据聚合的有效性; 2. 激酶偏好的生物学合理性:Top-1 显著预测主要集中在激酶家族(如 DDR1、FGFR1/2、KDR、RET、JAK1/2 等)。这在生物学上是合理的——激酶往往含有高度保守且构象刚性的 ATP 结合口袋,序列特征本身就能提供强烈的位点信号(以普拉替尼/Pralsetinib 为例,其靶向 11 种激酶的多激酶抑制活性正是基于这种保守性); 3. 消融实验的关键意义:序列-only 基线仅获得 4.8% 的显著案例(vs 完整流程的 31.7%),证明性能提升主要来自显式证据流水线(TM 拓扑、PTM 掩码、基序注释),而非 LLM 对序列模式的隐式记忆; 4. fpocket RCSB 近满分的解读:98.4% 的成绩源于 RCSB 共晶结构本身包含结合配体,口袋几何构象直接由配体定义,构成实质性的信息泄露,不可作为公平基线参考。
3.3 抗体表位评估(Group A,n = 26)
抗体表位评估面临的核心挑战是标注稀疏性:ABCD 数据库中大多数条目仅提供靶标和抗体名称,缺乏序列分辨率的残基级表位注释。最终筛选出 26 个有明确表位标注的案例(部分代表性例子见下表)。
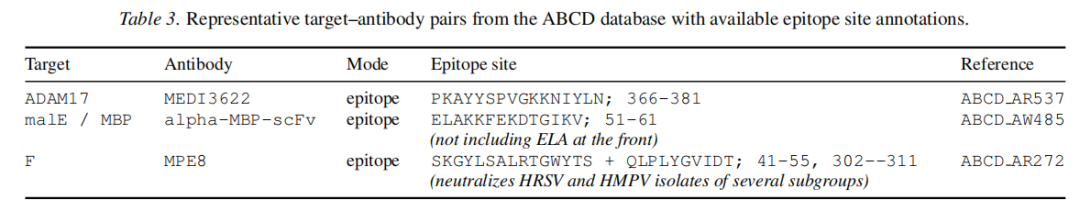
结果:Site4Drug 在表位模式下,Top-1 有 8/26 案例达到 p < 0.05 显著重叠,Top-5 达到 11/26,与口袋模式的显著率相近。考虑到表位注释的稀疏性和该数据集的噪声水平,这一结果具有现实意义。
3.4 结构可信度验证(pLDDT 分析)
尽管 Site4Drug 不直接使用蛋白质结构,研究者利用 AlphaFold3 对 63 个口袋验证案例的预测结构,检验预测位点的局部结构置信度(per-residue pLDDT):
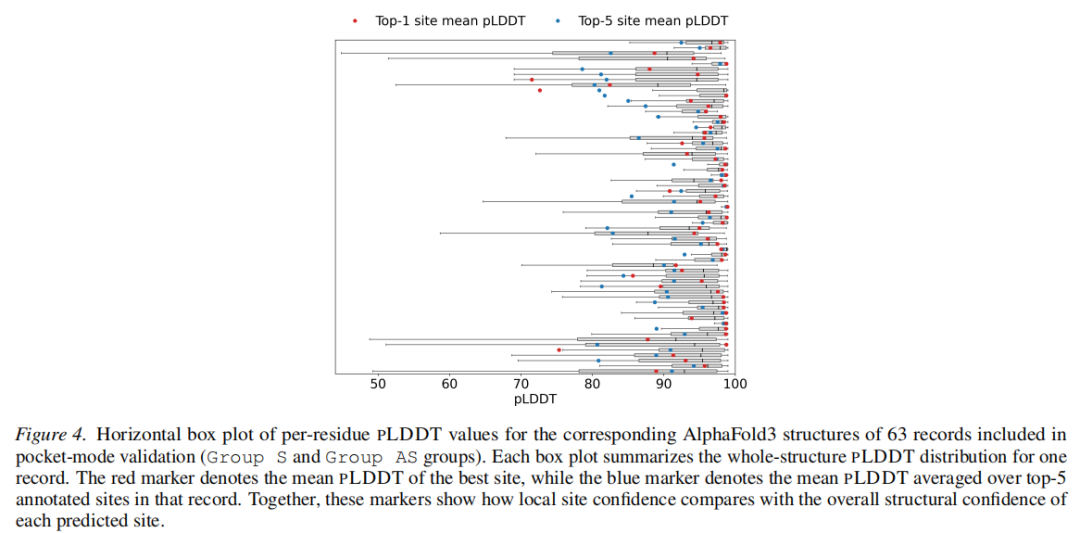
• 在大多数案例中,Top-1 预测位点的平均 pLDDT 高于 Top-5 均值(仅 9 个例外); • 这一规律表明:序列级证据聚合可能隐式地恢复了与结构可靠性相关的信号,尽管没有显式引入任何三维坐标信息。
该分析不仅是性能验证,更揭示了一个深层机制:蛋白质序列中的疏水性模式、保守基序分布、PTM 倾向性,本质上与结构稳定性和结合口袋的形成存在内在联系,而 Site4Drug 的证据流水线正是通过显式化这些信号,使得 LLM 能够捕捉到这种关联。
3.5 端到端演示(EGFR 案例研究)
口袋模式(DrugCLIP)
以 EGFR(RCSB: 1XKK,lapatinib 共晶)为例:
1. Site4Drug 从序列预测口袋区域(p 值 = 8.47 × 10⁻⁵); 2. 从 AlphaFold3 结构中提取该区域原子坐标,输入 DrugCLIP 虚拟筛选; 3. 已知 EGFR 抑制剂(afatinib、canertinib、erlotinib、gefitinib、lapatinib、osimertinib)均不在 DrugCLIP 数据库中; 4. 尽管如此,Top-6 命中化合物在结构上与已知抑制剂高度相似(共享苯环、环己烷等结合相关基团),结构重叠超几何检验 p < 10⁻¹¹; 5. 联合使用 Boltz2 进行结合亲和力验证。
表位模式(BoltzGen + AlphaFold3)
1. Site4Drug 预测 Top-5 EGFR 表位; 2. 通过 BoltzGen 为每个表位生成 50 条肽段候选(12–18 聚体); 3. 每个表位选取 BoltzGen 最优评分肽段,送入 AlphaFold3 做 5 次多聚体结构预测; 4. 计算中位 LIS(局部相互作用分数)和肽-靶标最近距离; 5. 结果与 Site4Drug 排名高度一致: • Rank-1(位点 44–98):胞外域,是唯一产生可定义 LIS 的候选(PAE < 12); • Rank-2(位点 348–358):对应 EGFR Domain III,与抗体药 petosemtamab、7D12 的已知靶向区域吻合; • Rank-5(位点 712–078):胞内区域,因 Site4Drug 正确识别其拓扑受限性而排名最末。
四、后训练实验:SFT 的双刃剑效应
论文坦率呈现了一个重要的负面结果。研究者以 Qwen3-235B Instruct 为骨干,在结构化 demonstration 上进行监督微调(SFT),观察到以下现象:
SFT 的正面效果:输出格式更规整,原因描述更简洁(平均 reason 长度从 20.6 词降至 5.5 词)。
SFT 的负面效果(捷径行为):
这一现象在 AI for Science 领域有普遍意义:格式监督可能与推理质量解耦。SFT 让模型"学会了如何写",却可能损害了"该写什么"的判断力。论文因此报告了基础模型的定量结果,并将生物学有意义的奖励信号(如基于真实 PTM 注释或已知结合位点的偏好对)视为靶点发现后训练的重要未来方向。
五、局限性的深入分析
论文对局限性的讨论相当克制且诚实,以下进行扩展解读:
5.1 四级结构盲区
当前 Site4Drug 以单链氨基酸序列为输入,但许多重要药靶(如离子通道、G蛋白偶联受体二聚体、整合素)以多亚基四级结构形式行使功能,其结合位点往往位于亚基界面。这一盲区限制了系统对同源/异源多聚体靶标的处理能力。
与 fpocket 和 RAPID-Net 不同(两者也仅处理单链),Site4Drug 的 LLM 核心原则上可通过扩展上下文(如提供多链序列及亚基组成描述)来处理这一问题,具备更好的可扩展潜力。
5.2 结构依赖性与 Token 瓶颈
口袋模式依赖结构上下文(需要 Module 2 的 AlphaFold3 结构);而 AlphaFold3 本身对超过约 5000 个氨基酸的序列不提供结构预测,这在处理超大蛋白时构成瓶颈。此外,将完整蛋白结构编码为 LLM 输入需要大量 tokens,不适合大规模筛选场景。
5.3 验证集规模与基线比较的公平性
评估数据集规模偏小(89 个靶标),特别是抗体表位组仅 26 个案例,统计功效有限。更严重的问题是数据泄露风险:RAPID-Net 等结构训练模型在 scPDB 数据集上训练,而 scPDB 来源于 RCSB,与验证数据存在显著重叠,因此被排除在基准比较之外。未来工作需要在严格数据分割控制下,与序列级或 apo 结构级 ML 基线进行公平对比。
5.4 浓度依赖性与脱靶效应
靶标接触呈现浓度依赖性:某些化合物在低浓度无活性,但在高浓度下可与特定靶标产生可测量的相互作用。Site4Drug 当前不整合浓度上下文,这在预测多靶标蛋白的优先成药区域时可能引入偏差。
5.5 二硫键建模精度
当前系统以半胱氨酸计数作为二硫键约束的代理,而非精确的二硫键配对预测。UNIPROT 中存在精确的二硫键注释,整合这一信息可显著提升含多个二硫键蛋白(如抗体、生长因子受体)的约束建模精度。
六、与相关工作的定位
6.1 Site4Drug 在工具链中的位置
蛋白序列
│
▼
┌─────────────────────────────┐
│ Site4Drug(本文) │ ← 上游瓶颈:选在哪里打?用什么打?
│ 约束感知靶点区域发现 │
└──────────────┬──────────────┘
│
┌────────┴────────┐
▼ ▼
表位模式 口袋模式
│ │
▼ ▼
BoltzGen DrugCLIP / BindCLIP
(肽/抗体生成) (小分子虚拟筛选)
│ │
▼ ▼
AlphaFold3 Boltz2
(多聚体验证) (亲和力评估)6.2 与 AI 科学智能体生态的关联
Site4Drug 的设计理念与近期 AI 驱动科学发现的智能体系统一脉相承:
| Site4Drug |
Site4Drug 的特色在于其决策可追溯性——不仅给出结果,还给出完整的推理链条,这在面向监管审查或实验设计归因时尤为重要。
总结与展望
核心贡献
Site4Drug 对靶点区域选择问题做出了以下方向性贡献:
1. 问题重新定义:将"选哪里"从直觉操作转化为约束优先的结构化决策问题,并通过完整审计日志实现可追溯性; 2. 模态自适应:无需用户预先指定药物类型,同一证据框架支持小分子、抗体、肽药三种模态的统一评估; 3. 序列级可行性:在不依赖结构的条件下实现与结构级基线相近的口袋预测性能,对新靶标场景(无结构可用)尤为重要; 4. 端到端集成:提供了从序列 → 靶点区域 → 分子设计的完整流水线接口,已与多个主流下游工具验证集成; 5. 失败归因能力:审计日志使"筛选失败是结合模型问题还是位点选择问题"这一历史难题变得可分析。
未来方向
• 生物学奖励信号驱动的后训练:构建基于 PTM 注释、已知结合位点、HDX-MS 数据的偏好对,支持更高质量的 RLHF 或 DPO; • 四级结构支持:扩展多链输入处理能力,覆盖通道蛋白、整合素等多亚基靶标; • 实验室闭环框架(Lab-in-the-Loop):整合实际质谱数据,实现 Site4Drug 预测-实验验证-迭代更新的主动学习循环; • 集成到自动化发现流水线:与 The Virtual Biotech(Zhang et al., 2026)等多智能体治疗发现框架的深度整合; • 大规模基准与公平比较:在严格的数据泄露控制下扩大验证规模,并纳入序列/apo结构级 ML 基线。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢