
DRUGONE
蛋白质是绝大多数生物学过程的直接执行者,因此蛋白质组学对于理解疾病机制和细胞功能具有不可替代的重要价值。然而,相较于RNA测序技术,蛋白质定量检测成本更高、覆盖范围更有限,尤其是在IDH突变型胶质瘤等疾病研究中,大规模蛋白质组数据十分匮乏。虽然利用转录组数据推断蛋白质丰度具有重要意义,但RNA表达与蛋白质丰度之间通常仅存在较弱相关性,使这一任务长期面临挑战。
研究人员开发了一种整合多组学信息的深度学习框架T2Pdecoder,通过学习RNA与蛋白质之间共享的潜在表示空间,实现大规模蛋白质丰度预测。该模型能够预测5738种蛋白质的表达水平,并同时适用于Bulk RNA测序和单细胞RNA测序数据。
在多个胶质瘤队列以及乳腺癌队列中的评估结果显示,T2Pdecoder预测得到的蛋白质谱比原始RNA表达更接近真实蛋白质组学结果。同时,在通路富集分析、肿瘤亚型识别、生存分层以及单细胞状态解析等多个任务中,T2Pdecoder均展现出优于传统RNA分析的能力。研究表明,该方法能够利用现有海量转录组数据开展蛋白质中心分析,从而获得传统RNA分析难以揭示的生物学信息。

胶质瘤是成人最常见的恶性脑肿瘤之一,尽管近年来手术、放疗和化疗不断进步,但患者总体预后仍然较差。深入理解胶质瘤发生发展的分子机制,对于发现新的诊断标志物和治疗靶点至关重要。
过去十余年中,大规模转录组测序推动了肿瘤生物学研究的发展。然而,RNA表达只能反映基因转录水平,而蛋白质才是绝大多数细胞功能的直接执行者。大量研究已经证明,RNA丰度与蛋白质丰度之间往往存在较弱相关性,因此仅依赖RNA分析难以准确刻画真实的功能状态。
近年来,CITE-seq等多组学技术实现了RNA与蛋白质的同步测量,并推动了RNA到蛋白质预测模型的发展。但现有方法主要存在两个问题:其一,大多数模型只能预测几十到数百种蛋白质;其二,模型主要针对单细胞数据设计,难以推广到临床常见的大规模Bulk RNA数据。
研究人员因此提出T2Pdecoder,希望建立一个能够同时适用于Bulk和单细胞数据、支持全蛋白质组尺度预测的统一框架,从而充分利用大量已有转录组资源开展蛋白质层面的功能研究。
方法
T2Pdecoder采用迁移学习与多组学融合策略构建。首先利用CPTAC泛癌种配对RNA-蛋白质数据训练CLIP模型,通过两个独立编码器分别学习RNA表达模式和蛋白质丰度模式,并建立统一的跨模态嵌入空间。随后利用大规模蛋白质组数据训练多任务变分自编码器,从仅有蛋白质数据的样本中学习蛋白质丰度分布规律。最终,将RNA编码器与蛋白质解码器整合,构建从全转录组表达谱直接预测5738种蛋白质丰度的生成框架。该模型既可应用于Bulk RNA测序数据,也可扩展至单细胞RNA测序数据,从而支持蛋白质中心的功能分析、亚型识别和细胞状态解析。
结果
T2Pdecoder总体框架与模型构建
研究首先构建了T2Pdecoder整体框架。模型利用1351个泛癌种RNA-蛋白质配对样本学习跨模态共享表示空间,再结合1422个胶质瘤蛋白质组样本训练蛋白质生成模块。
在CLIP训练阶段,来自同一样本的RNA和蛋白质嵌入向量表现出高度一致性,而不同样本之间则保持明显分离。进一步在胶质瘤数据集上的微调结果显示,这种跨模态映射关系能够稳定迁移到新的队列中。
研究同时发现,目前可获得的胶质瘤蛋白质组数据高度偏向IDH野生型,而IDH突变型胶质瘤蛋白质组数据十分有限。T2Pdecoder的出现为这些缺乏蛋白质组数据的亚型提供了开展蛋白质层面研究的可能性。
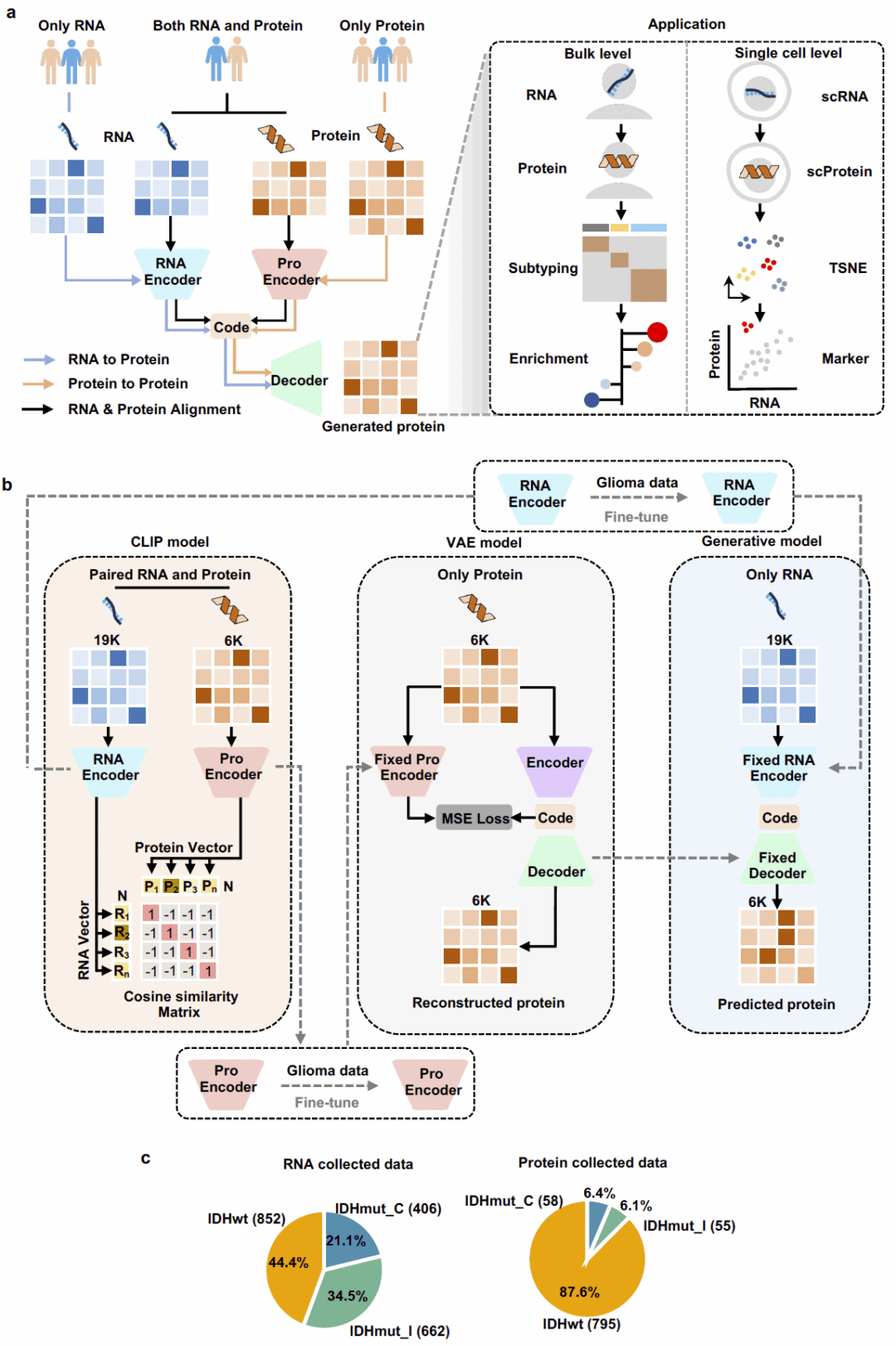
图1: T2Pdecoder整体框架示意图。
蛋白质丰度预测性能评估
研究利用具有RNA和蛋白质双重测量的数据集对模型进行了系统验证。结果显示,T2Pdecoder预测得到的蛋白质丰度与真实蛋白质组学测量结果保持稳定相关性,并且在训练集、验证集以及独立外部测试集上均保持一致表现。
进一步分析发现,模型最准确预测的蛋白质主要集中在细胞增殖和氧化磷酸化相关通路中。这些通路也是胶质瘤生物学研究中的关键功能模块。
研究随后将模型扩展至乳腺癌数据集。尽管训练样本数量较少,但模型仍然能够保持与胶质瘤相似的预测性能,并成功重现细胞增殖和氧化磷酸化等核心生物学通路。
这些结果说明,T2Pdecoder具有良好的跨癌种迁移能力和泛化能力。
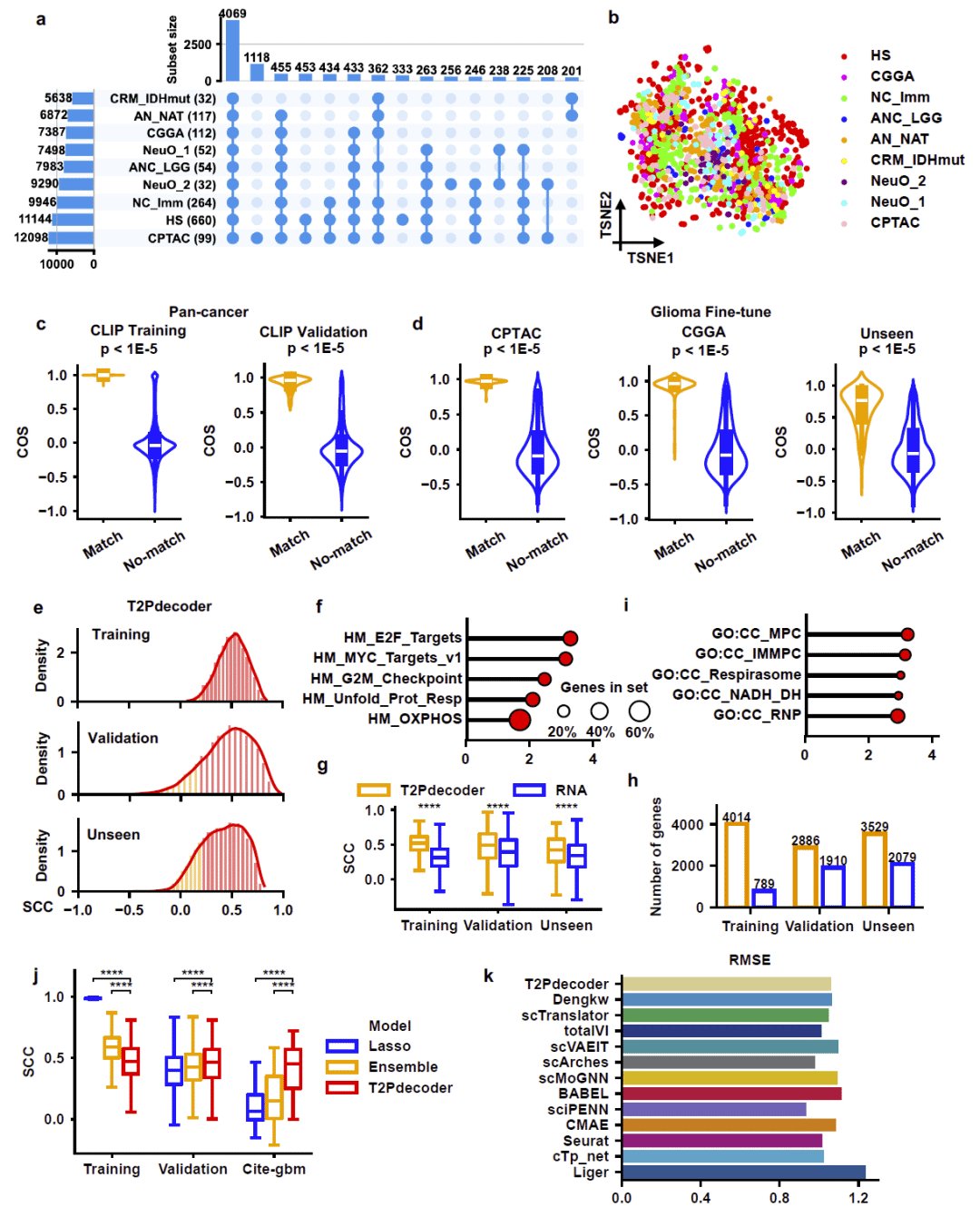
图2: T2Pdecoder蛋白质预测性能评估。
与传统RNA分析的比较
研究进一步评估T2Pdecoder是否优于直接将RNA表达作为蛋白质替代指标。
结果显示,在绝大多数基因上,T2Pdecoder预测得到的蛋白质丰度与真实蛋白质水平之间的相关性均高于RNA本身。即使对于RNA与蛋白质天然相关性较弱的基因,T2Pdecoder仍然能够显著提高预测准确度。
研究同时比较了Lasso回归和随机森林等传统机器学习模型。虽然这些模型在训练集上表现良好,但在独立验证集和单细胞测试集上出现明显过拟合。而T2Pdecoder在不同数据集间保持稳定性能,展现出更好的泛化能力。
在单细胞CITE-seq数据集上,研究进一步与11种主流RNA-蛋白质预测算法进行了比较。结果表明,T2Pdecoder整体性能处于第一梯队,并且能够预测远多于现有方法的蛋白质数量。
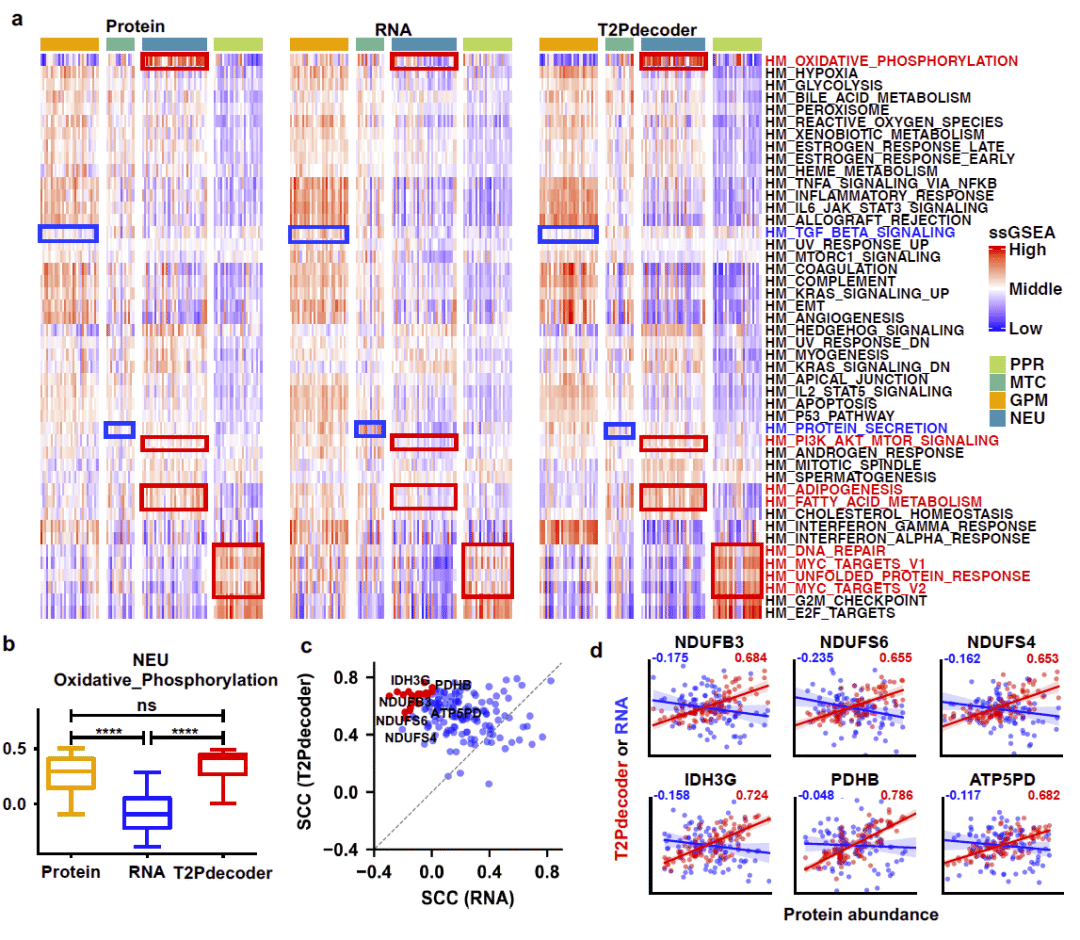
图3: T2Pdecoder与传统RNA分析及现有预测方法的比较。
蛋白质层面的功能通路解析
研究随后评估T2Pdecoder是否能够重建真实蛋白质组学中的功能状态。
在CPTAC胶质瘤队列中,研究分别利用真实蛋白质组、RNA表达谱以及T2Pdecoder预测蛋白质谱进行通路富集分析。结果发现,炎症反应和干扰素信号等通路在三种数据中均可观察到。然而,氧化磷酸化、脂肪酸代谢以及细胞周期等关键代谢通路,仅在真实蛋白质组和T2Pdecoder预测蛋白质组中呈现一致富集,而RNA表达分析无法准确重现这些模式。
以氧化磷酸化通路为例,研究发现T2Pdecoder预测结果与真实蛋白质组之间高度一致,而RNA表达则无法反映该功能状态。这些结果说明,T2Pdecoder能够恢复RNA分析所丢失的重要功能信息,从而更接近真实蛋白质组学层面的生物学状态。
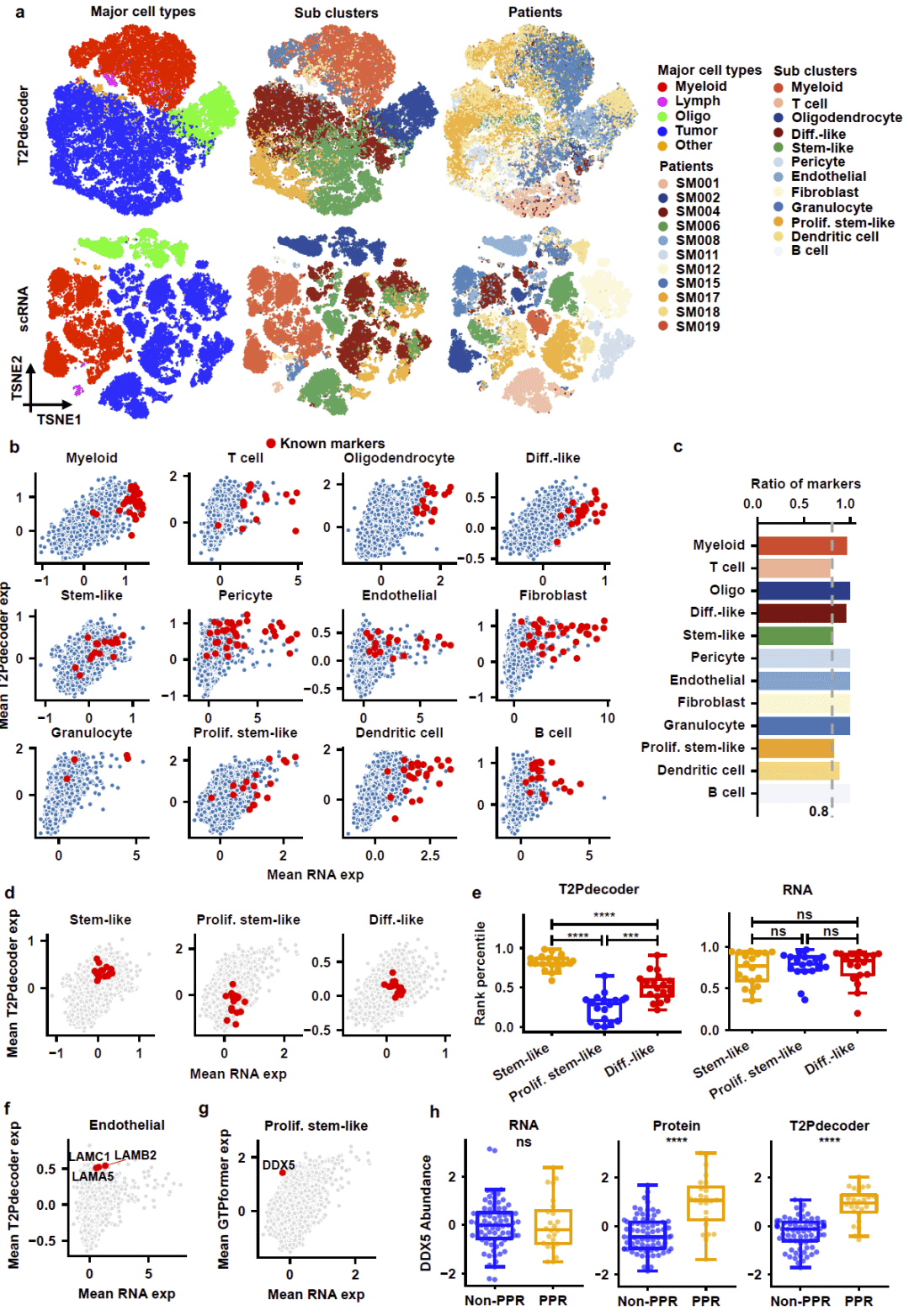
图4: T2Pdecoder揭示蛋白质层面的功能通路特征。
单细胞与胶质瘤亚型分析
研究进一步将T2Pdecoder应用于单细胞RNA测序数据和大规模胶质瘤队列分析。
在55284个单细胞组成的胶质瘤图谱中,T2Pdecoder预测得到的蛋白质表达谱能够准确区分不同细胞类型和细胞亚群。同时,预测蛋白质谱明显减弱了患者间批次效应,提高了生物学信号的可解释性。
研究发现,在肿瘤干细胞状态转换过程中,氧化磷酸化相关蛋白质呈现明显动态变化,而这些变化在RNA表达层面几乎无法观察到。此外,DDX5等促进肿瘤增殖的重要蛋白在预测蛋白质层面表现出显著差异,但其RNA表达并不显著。
随后,研究利用1920例胶质瘤样本开展蛋白质层面的无监督聚类分析。结果识别出四个稳定的功能亚群,这些亚群在所有分子亚型中均保持一致,并表现出显著不同的生存结局。其中两个亚群预后较好,而另外两个亚群与更差生存期显著相关。多因素分析进一步证明,这种蛋白质亚型分层具有独立于肿瘤分级之外的预后价值。
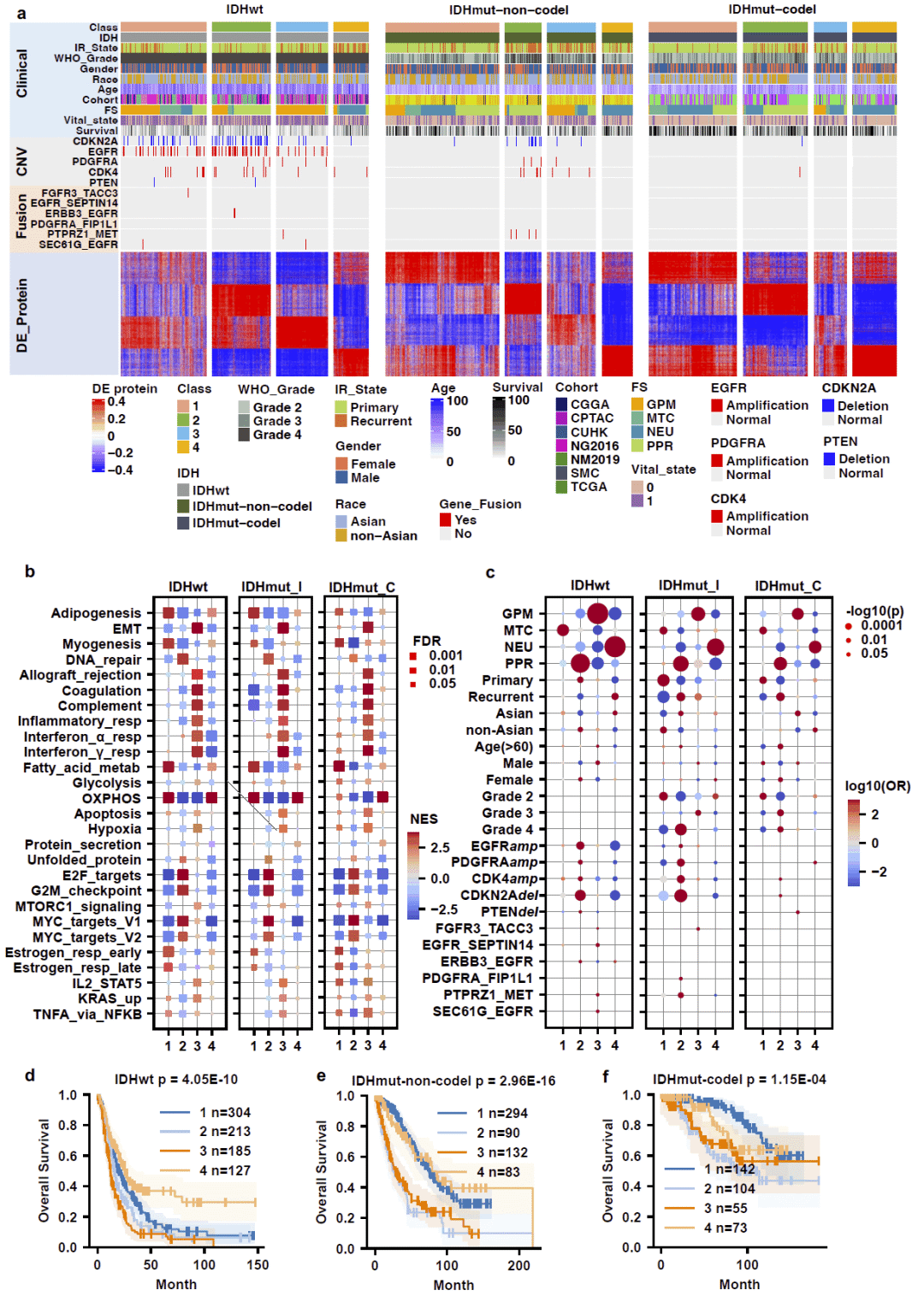
图5: 基于T2Pdecoder预测蛋白质组的单细胞分析与胶质瘤分型。
讨论
本研究提出的T2Pdecoder建立了一种从转录组到蛋白质组的大规模推断框架,为缺乏蛋白质组数据的研究场景提供了新的解决方案。
与现有方法相比,T2Pdecoder最大的特点在于不依赖基因与蛋白质的一一对应关系,而是通过共享潜在空间学习RNA与蛋白质之间复杂的多对多映射关系。同时,该模型能够预测5738种蛋白质,远超现有大多数RNA到蛋白质预测模型仅支持几十至数百种蛋白质的规模。
研究结果表明,即便蛋白质丰度预测精度提升幅度有限,模型依然能够在功能通路分析、肿瘤分型和单细胞状态解析等下游任务中恢复大量蛋白质层面的生物学信息。这说明蛋白质中心分析能够提供超越传统RNA分析的重要补充视角。
特别是在单细胞分析中,T2Pdecoder揭示了氧化磷酸化代谢重编程和DDX5相关增殖信号等关键特征,而这些现象在RNA层面并不明显。与此同时,在大规模胶质瘤队列中,模型预测的蛋白质组能够构建具有独立预后价值的功能分型体系,进一步证明了其临床应用潜力。
研究人员指出,目前模型主要在胶质瘤和乳腺癌中进行了验证,未来随着更多RNA-蛋白质配对数据积累,该框架有望推广至更多癌种和正常组织。此外,未来还可进一步整合DNA甲基化、空间转录组以及病理图像等多模态信息,构建更加全面的多组学解析平台。
总体而言,T2Pdecoder展示了一种利用现有海量转录组资源开展蛋白质组学研究的新范式,为癌症研究和精准医学中的蛋白质中心分析提供了重要工具。
整理 | DrugOne团队
参考资料
Wang, H., Tang, J., Qiao, Y. et al. T2Pdecoder enables protein-centric analyses from transcriptomic data. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-74209-3

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢