
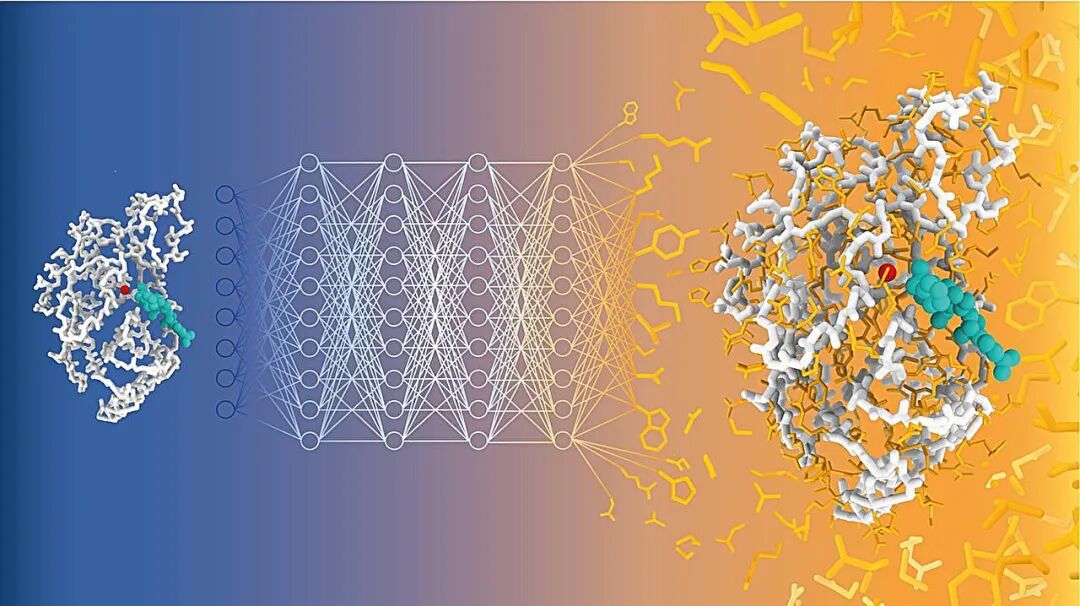
蛋白质相互作用建模是蛋白质设计的核心,而机器学习已彻底改变了蛋白质设计,并在药物发现及其他领域得到了广泛应用。在此背景下,基于结构的从头结合剂设计通常被分为条件生成建模和通过结构预测器进行序列优化(“幻觉”)。我们认为这是一种错误的二分法,并提出了 Proteina-Complexa,一种全新的全原子结合剂生成方法,它统一了这两种范式。我们扩展了近期基于流动的潜在蛋白质生成架构,并利用单体计算预测蛋白质结构的结构域间相互作用构建了 Teddymer,这是一个用于预训练的大规模合成结合剂-靶标对数据集。结合高质量的实验多聚体,这可以训练出一个强大的基础模型。然后,我们利用这种生成先验进行推理时优化,从而融合了先前不同的生成方法和幻觉方法的优势。 Proteina-Complexa 在计算结合剂设计基准测试中树立了新的标杆:其计算机模拟成功率显著高于现有的生成式方法,并且我们新颖的测试时优化策略在标准化的计算预算下远胜于以往的模拟方法。此外,我们还展示了界面氢键优化、折叠类别引导的结合剂生成,以及对小分子靶标和酶设计任务的扩展,再次超越了以往的方法。

论文:Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
单位:NVIDIA、牛津大学、Mila、蒙特利尔大学、CIFAR、AITHYRA、首尔大学
发布日期:2026
Code:
https://research.nvidia.com/labs/genair/proteina-complexa/
请索引第104篇论文
 |  |
给AI+生命科学交叉研究者的核心启示:蛋白binder设计中长期对立的两大学派——"生成派"vs"幻觉派"——的本质矛盾,其实在LLM领域早已被回答了。答案不是选边站,而是像o1/R1那样,把预训练的generative prior和inference-time search融进同一个框架里。
01 为什么难
蛋白binder(结合蛋白)设计是计算生物学皇冠上的明珠之一——设计出能高亲和力结合特定靶点(某个体表蛋白、病毒刺突、自身抗原…)的小型蛋白,本质上就是在做 de novo药物发现:比抗体更小、更稳定、更可工程化、更低生产成本。
但难点也是结构性的:
蛋白相互作用由3D结构介导,你必须同时搞定序列(20种氨基酸的离散选择)和全原子坐标(连续空间几何),且两者强耦合。
实验PDB里的天然多聚体(multimer)数据只有~22.5万条目,过滤完高质量链间界面的可用binder-target对可能就几千条——对一个需要学通用规律的深度生成模型而言,数据严重不足。
传统方法分裂成两个看似不相容的世界观:
生成式方法(RFDiffusion等) | 幻觉方法(BindCraft等) | |
|---|---|---|
思路 | 在binder-target复合物结构上训练条件生成模型,推理时从噪声采样新binder | 固定目标结构,用AlphaFold2的置信度/ipAE等作为奖励信号,对序列做梯度优化 |
优点 | 学得数据分布先验,采样相对快,多样性好 | 不需要训练数据(只需要折叠器),直接优化最终评判指标 |
致命伤 | 数据饥荒 → 先验不够好;生成后要ProteinMPNN重设计序列 | 无生成先验 → 从随机/差起点开始优化;gradient through discrete sequence需要ad-hoc松弛;计算贵且不稳定 |
如本文作者一针见血指出的——这就像LLM时代之前:要么纯训练时优化(SFT),要么纯推理时搜索但不懂语言(早期符号推理)。而o1/R1/STaR等的启示恰恰在于:预训练base model × inference-time compute scaling = 最佳组合拳。
本文提出的 Proteina-Complexa(Complexa) 做的正是这件事——第一次将flow-based全原子生成先验与test-time search统一在同一个binder设计框架中。
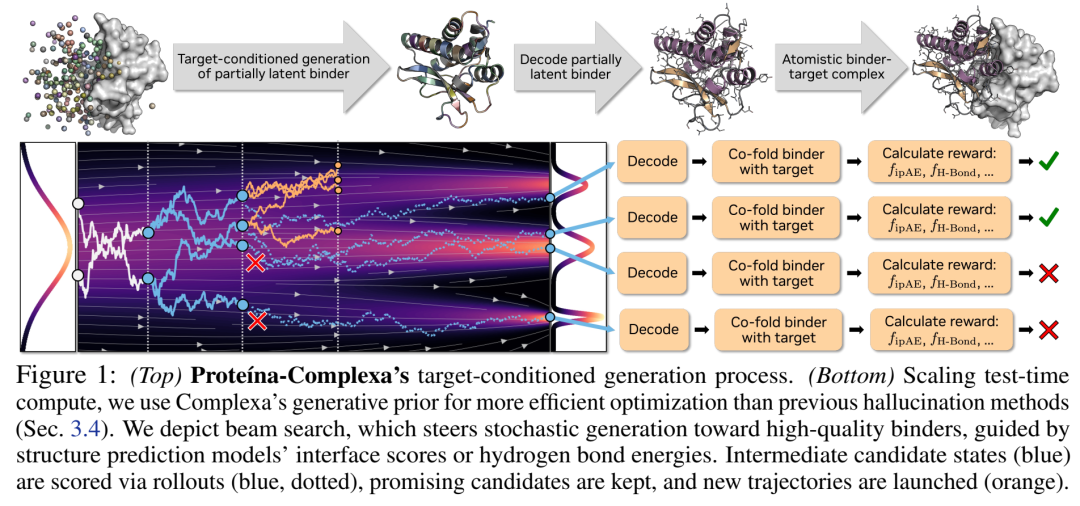
02 核心架构:La-Proteina → Complexa 是如何扩展的
2.1 La-Proteina的"部分隐变量Flow Matching"范式
先把底座讲清楚。La-Proteina(Geffner et al., ICLR 2025/2026)是一个全原子蛋白生成模型,它的聪明之处在于"分而治之"的混合表示:
Cα坐标→ 显式建模(连续)
其余一切(侧链原子坐标 + 氨基酸类型)→ 压缩进逐残基的连续隐变量 ,由一个VAE学出来
即所谓 partially latent representation:序列信息和全原子细节被编码到固定大小的per-residue latent里,生成模型只在 这个连续空间中做 rectified flow matching。解码时VAE decoder从 重建出全原子结构+序列。
关键优势:
把混合离散/连续、变长的复杂生成问题变成固定维度连续空间的flow问题
Cα和z用不同schedule去噪(Cα更快→先粗后细),高效
不用triangular multiplicative/update层(不像AF2),纯transformer with pair-biased attention → 快
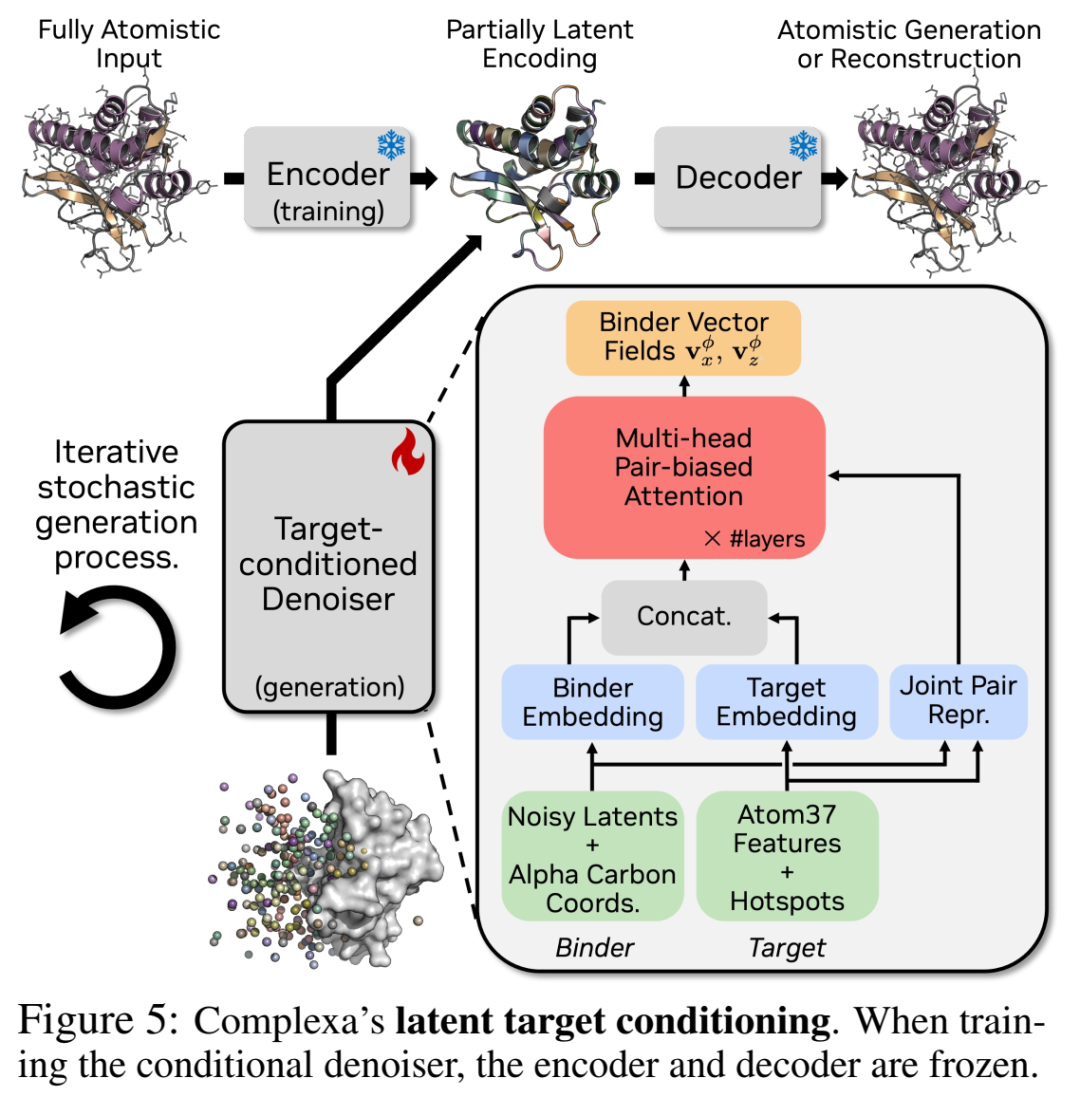
2.2 扩展到binder设计:Latent Target Conditioning
Complexa的核心修改非常干净——
VAE只管单体binder的编解码(不变)。只有flow model变成条件生成:输入多出target的表征 ctarget,输出仍只生成binder。
具体来说(对应论文Fig.5 / Sec. 3.2):
Target用 Atom37表示(每残基最多37个原子坐标 + aa identity one-hot)+ binary hotspot token(标记界面附近的关键残基)
这些target embedding沿sequence dim拼接到binder的 后面
同时构建 joint pair representation:binder内部pair + target内部pair + binder-target cross pair(靠Cα-backbone距离等)
Transformer denoiser在这个extended序列+pair上做pair-biased attention
一个极其重要的细节:他们在插值中加入了 translation noise,扰动binder全局质心:
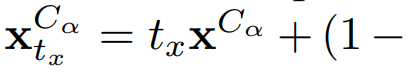
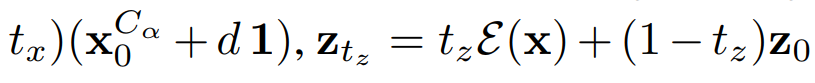
直觉:普通flow model最低频模式=全局平移,如果不加这个noise,模型会"走捷径"——记住target在原点的绝对位置而不是学相对放置。加了之后强制模型在整个去噪过程中持续refine全局定位。消融实验(表6/表7)证实了这一步的关键性。
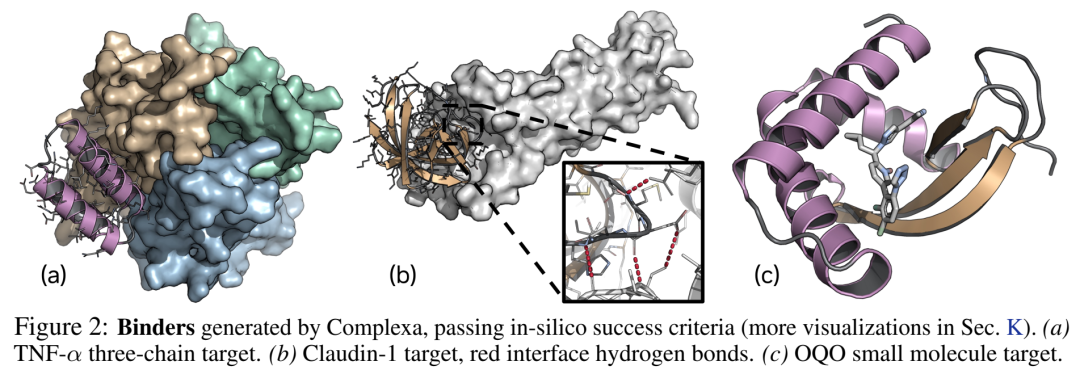
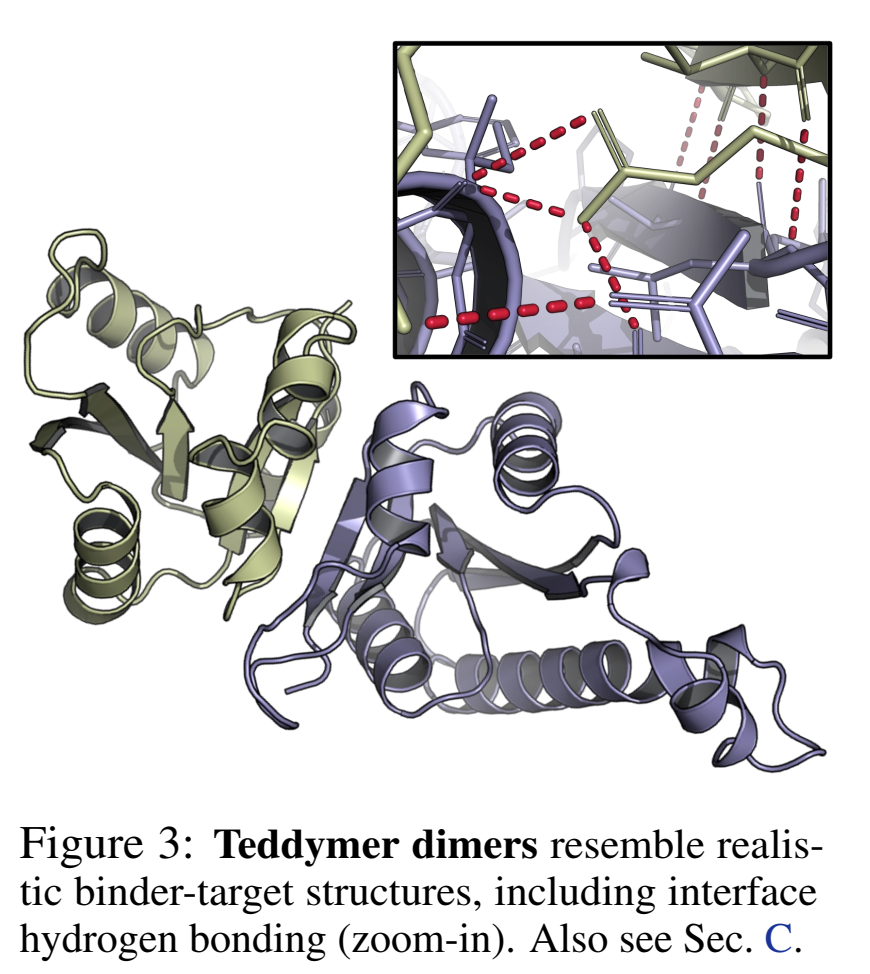
03 数据饥荒怎么破:Teddymer —— 从AFDB域间相互作用造出千万级合成训练数据
这是论文最实用的贡献之一。
问题:PDB里靠谱的binder-target dimer/多聚体太少(过滤完就~4.6万条)。
洞察(Sec 3.1):AlphaFold DB有2亿+单链预测结构,其中很多是多域蛋白。如果用 TED(The Encyclopedia of Domains) 的结构域注释把每个多域单体切成独立域,再把域之间空间邻近的配对抽出来当"人工二聚体"——它们的域-域界面在生物物理上类似于真实链-链界面。
Pipeline:
从AFDB50(clustered版,4718万结构)取含TED注释的条目
按域拆链 → 抽所有二域对,筛选CA-CA距离<10Å的残基≥4对 → 1008万个二聚体
用Foldseek-Multimer按链级结构相似+界面相似聚类 → 355万个簇
再过滤 interface-pLDDT>70, ipAE<10, interface length>10 → 最终训练用cluster rep ~51万条(整体~711万条带非rep)
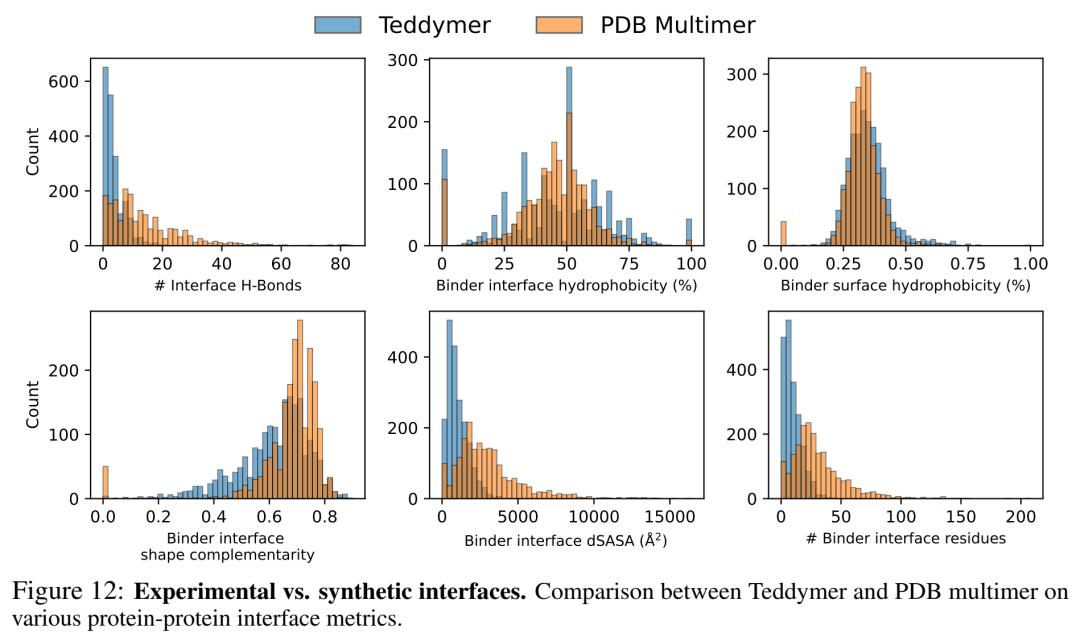
论文Fig.12对比了Teddymer界面vs PDB界面在氢键数、疏水性、形状互补性等6个metric上的分布——显著重叠但有额外多样性,说明这是合理的data augmentation而非纯噪声。
消融:
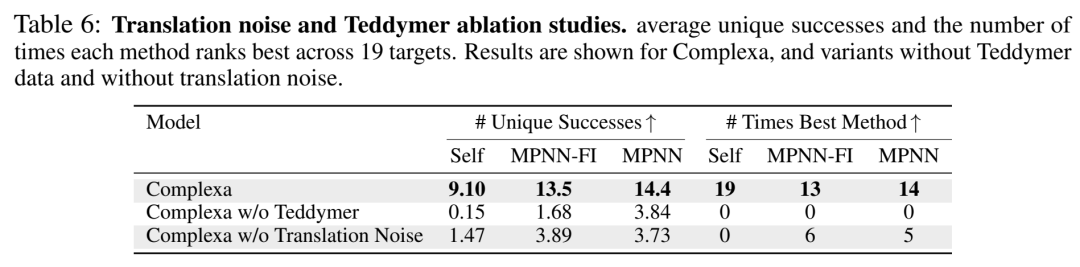
去掉Teddymer后,Self序列成功从9.10暴跌到0.15——practically unusable。这说明PDB-only数据量根本撑不起这个条件生成任务的泛化。而且作者还做了额外验证(I.1.1):即使换用RF3或Boltz-2做评估而非AF2-Multimer,Teddymer的收益仍然robust——排除"因为AF2既是数据来源又是评估器所以虚高"的质疑。
💡 对AI+跨域的启示#1:当你面临domain-specific高质量数据极端稀缺时,"合成数据的合理性"不靠理论证明,靠(1)biophysical plausibility论证 + (2)严格消融 + (3)换independent evaluator做sanity check。Teddymer这套recipe(从大规模预测结构中抽取子结构关系→过滤→聚类去冗)其实是通用的。
04 训练策略:三阶段,仿大模型pretrain→finetune管线
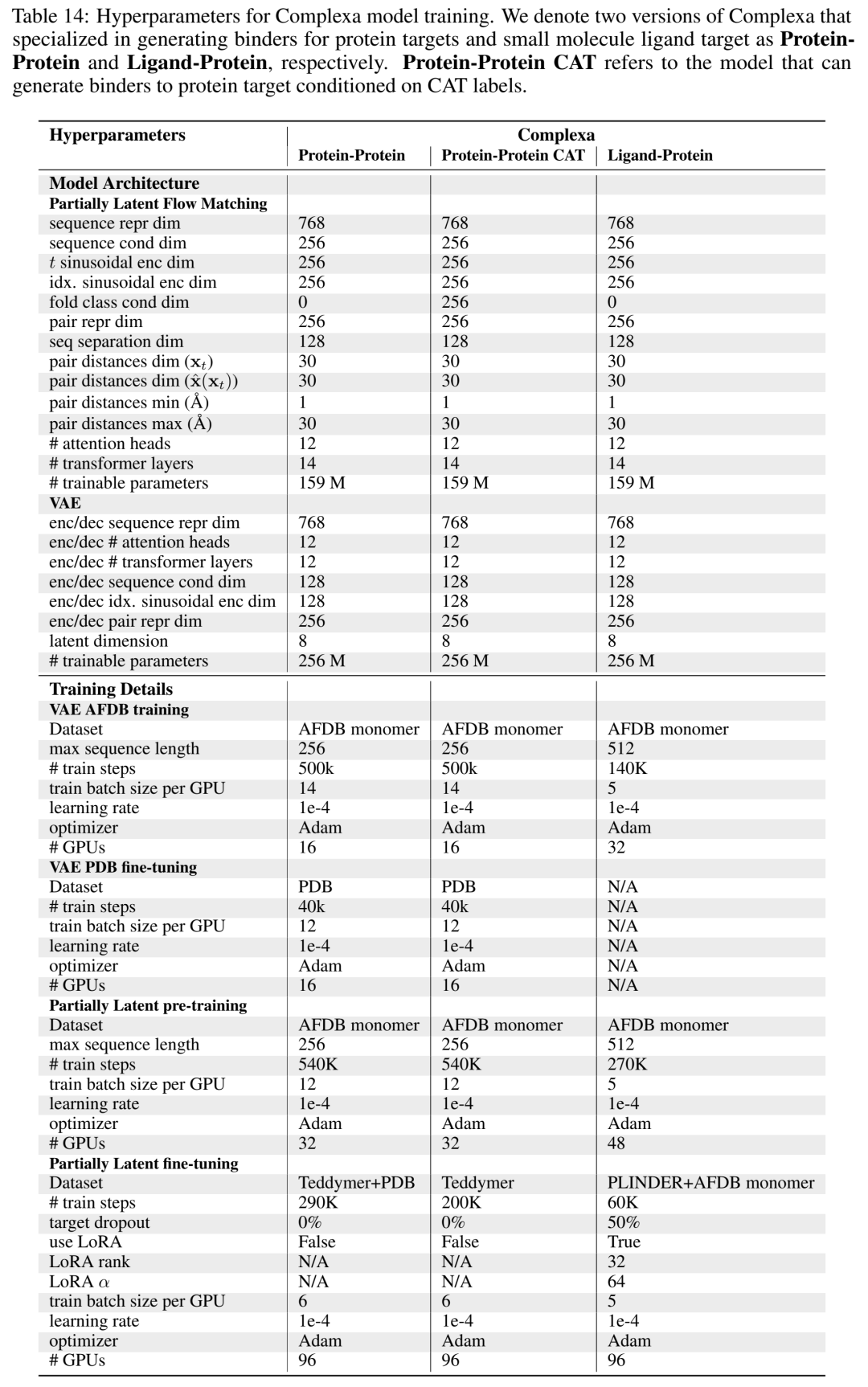
小分子版多了一步:因PLINDER太小,用LoRA做轻量conditioning finetune,且50%概率mask掉配体特征防过拟合。
这种staged protocol的设计哲学很明确:先让模型懂"什么是合理蛋白",再教它"怎么对着target摆一个binder"——和LLM的continue pretraining→SFT→(可选)RL的逻辑同构。
05 Test-Time Compute:把Inference-Time Search搬进蛋白设计
这才是论文最"LLM o1风格"的部分(Sec 3.4)。
5.1 四种搜索算法
算法 | 核心思想 | 代价特征 |
|---|---|---|
Best-of-N | 增大N,采样越多越可能撞到高ipAE样本,筛成功者 | 纯并行,但浪费——盲目采样 |
Beam Search | 维护N条轨迹,每K步分支L次→rollout全清→按reward选top-N推进 | 结构化探索,K=100时每轮decode+fold N×L个候选 |
Feynman-Kac Steering | 同上但不deterministic top-N,而是按exp(β·R)重要性采样 → 粒子滤波式倾斜分布 | 更soft,避免beam search的mode collapse倾向 |
MCTS | 把去噪过程当树:selection(UCB)→expansion(跑K步)→rollout→backprop | 理论上最优explore-exploit折中,但continuous action space要改造成stochastic expand |
关键工程决策:他们不做Tweedie denoised estimate of reward(中间态太noisy不可信),而是roll out每条候选到clean sample再decode+fold算reward——因为Complexa的生成足够快(~15s/sample),fully denoising的cost可接受。
Reward就用结构预测器的指标:主要是 (inter-chain predicted Aligned Error),越低越好。也可叠加氢键能量 。
5.2 结果:Search碾压纯幻觉方法
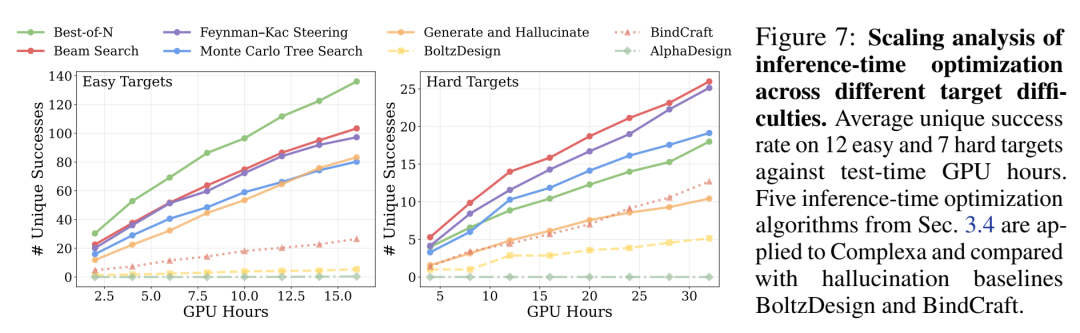
图7(核心图) 是他们放的最有说服力的证据——横轴GPU小时,纵轴平均unique success rate,分easy target(12个)和hard target(7个):
Easy targets:Best-of-N已经干翻BindCraft/BoltzDesign/AlphaDesign
Hard targets:Beam Search / FKS / MCTS显著拉开差距
BindCraft在同等算力下要么产出少(反复backprop through AF2慢),要么撞不上有效区域
💡 对AI+跨域的启示#2:这里的本质lesson是——当一个领域有(1)一个还行的generative prior + (2)一个可快速evaluate的reward proxy,test-time compute scaling就会work。关键瓶颈往往不在search算法本身,而在(1)prior够不够好(Teddymer就是干这个的)和(2)evaluator够不够cheap(所以他们死磕效率→不用triangular layer→纯transformer→15s/样)。
06 基模型 + 端到端全原子序列都打SOTA
表2 蛋白target基准(200样/靶,无inference-time search)
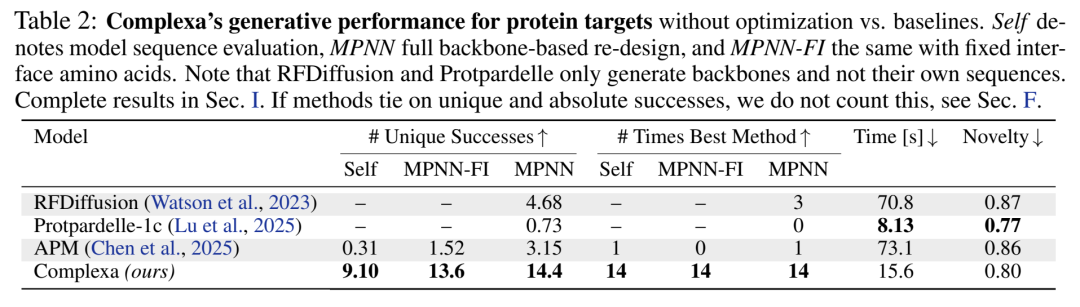
三条结论同时成立:
Complexa的自生成序列(Self)就已经超过baseline用ProteinMPNN重设计后的结果 → 说明它真的在学co-design(序列+结构一起),而不是只画了个好看的backbone然后外包给MPNN
速度快约 4.5× vs RFDiffusion、4.7× vs APM
Novelty(TM-score对PDB的距离)保持合理水平~0.80,没有坍缩成memorize训练集
表1 小分子target(SAM/OQO/FAD/IAI)

唯一公开的全原子小分子binder生成baseline被全方位超过。
酶设计(AME benchmark, 41 tasks)
Method | Success (Unique / 41) |
|---|---|
RFDiffusion2 + LigandMPNN(8) | 30 |
Complexa + Self-generated seq | 41 |
Complexa + single LigandMPNN | 40 |
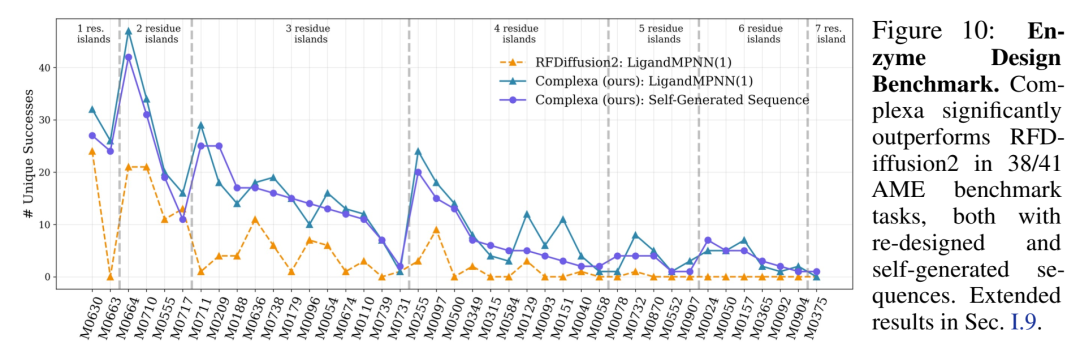
Fig.10的柱状图对比非常直观——Complexa在困难的多岛催化残基任务上也稳定赢。
07 两个很酷的"附加技能"
7.1 界面氢键优化
他们证明了reward可以不止是黑盒ipAE——直接把Rosetta的interface H-bond count/energy纳入beam search reward:
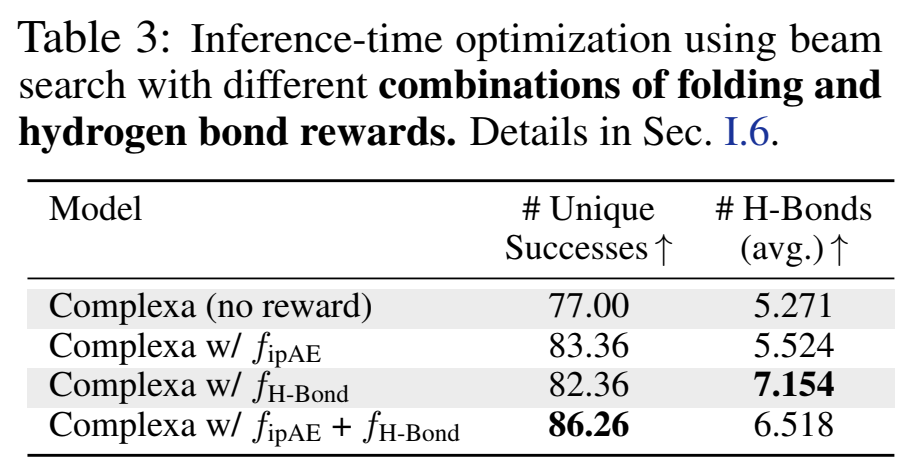
氢键数从5.3拉到7.1,且联合reward还能再抬成功数。更重要的是,ipAE和H-bond数呈Spearman -0.69负相关——优化前者不会adversarially牺牲后者,反而是协同的。这对"可解释奖励/物理奖励混用"的方向很有启发。
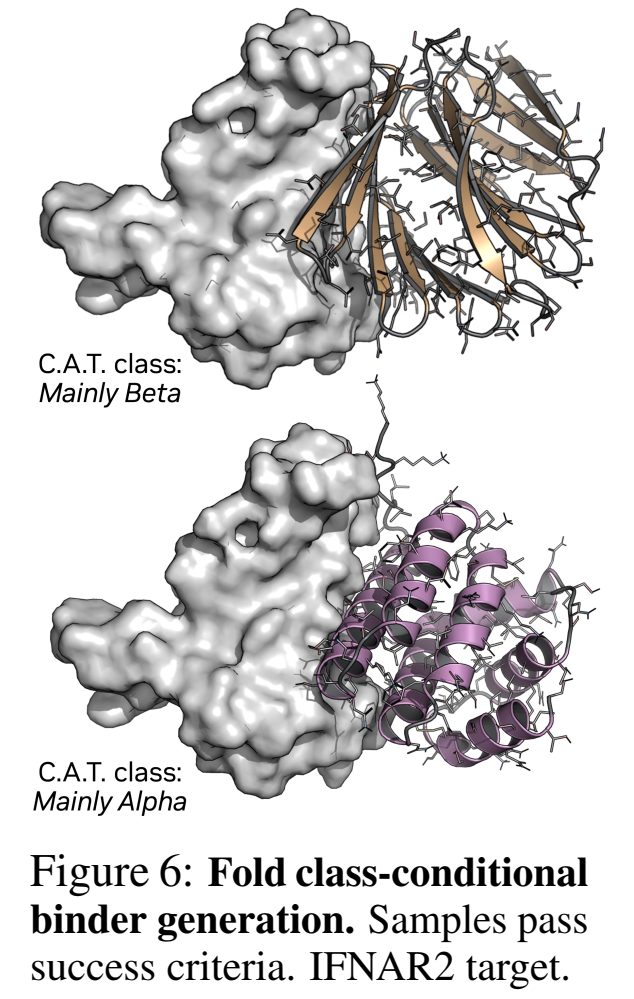
7.2 Fold Class引导:打破"全是α螺旋"的偏见
以前的生成器容易bias α-helical(因为PDB里螺旋多+扩散容易卡在简单流形)。他们在Teddymer上训了带CATH C-level标签(Mainly-Alpha / Mainly-Beta / Mixed)条件的版本,可以在推理时指定你要β-sheet为主的binder还是α为主的——直接拓宽了化学空间探索范围。
08 这篇文章强在哪
维度 | 评价 |
|---|---|
概念整合力 | 把"生成 vs 幻觉"的对立框架性消解——不是第一篇用flow做binder,但是第一篇系统性把inference-time scaling作为一个budget-controllable dial引入并benchmarks |
数据工程 | Teddymer的构造有理有据,消融铁证,还做了cross-folder sanity check |
端到端全原子 | 序列+结构一起出,Self就work,减少pipeline fragility |
效率 | 159M denoiser参数,无triangular layer,~15s/sample——这是能做search的前提 |
诚实度 | 消融有反面结果(w/o Teddymer崩了→他们如实show而非hide),limitation节承认缺湿实验验证 |
需要审慎对待的点
全是in-silico metrics:ipAE/pLDDT是proxy,不是Kd/ΔG实验。论文自己也写了future work需要wet lab validation——这一点读者务必清醒。
Teddymer来自AF2预测结构:虽然cross-folder check缓解了担忧,但根本上所有"用预测结构训练→用预测结构评"都有distributional闭路风险。不过坦率说——这不是本文独有的问题,是整个field的现状。
Hard target上BindCraft偶尔反超(如SC2-RBD、BetV1)——提醒我们generative prior不是万能药,尤其当训练分布没覆盖到时
PLINDER太小 → LoRA finetune的策略对小分子binder泛化能力的上限还需更多target验证
09 对你(AI+交叉学研究生)的三个takeaway
① "Pretrain + Test-Time Compute"不只是LLM的故事
本文证明同一模板在3D分子生成同样成立:pretrain给先验(flow model on Teddymer+PDB),test-time compute给定向改进(beam/MCTS on ipAE reward)。如果你在做其他科学生成的domain——晶体结构?催化剂表面?材料微结构?——问自己同一个问题:你的field有没有一个"还行的生成先验"和一个"cheap-ish evaluator"可以hook search上去?
② 数据稀缺的解法:从大规模"第二手数据"里榨出结构关系
AFDB的2亿预测单体不是gold-standard,但域-域界面的关系层面的生物物理约束是transferable的。Teddymer的精髓是:不把预测结构当truth,而是把它当"提供足量界面样例让模型学界面几何规律"的训练脚手架。这种"关系级迁移 > 实例级保真"的思维在很多低数据科学domain都值得借鉴。
③ 效率不是事后优化——它是enable新范式的前提
没有~15s/sample的速度(来自non-triangular transformer + latent space紧凑性),Beam Search/MCTS根本跑不起来。论文里最容易被忽略的一句话其实是附录的denoiser规格:"14 layers, 159M params, no triangular updates"。当你设计ML for science pipeline时,问的不只是"它能work吗",而是"它能work fast enough that搜索/迭代/主动学习在经济的时间预算内?"
如有想深挖的具体模块(flow matching公式推导、pair-biased attention细节、MCTS在连续状态空间的实现陷阱、或者Teddymer的数据处理脚本逻辑)——欢迎评论区点题,后续可单独拆一期。



内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢