随着mRNA疫苗和治疗药物从概念验证走向多疾病应用,如何在mRNA序列中平衡翻译效率、稳定性、免疫原性和可制造性,成为下一代核酸药物设计的重要问题。与传统小分子或重组蛋白药物不同,mRNA药物的药效学和药代动力学性质很大程度上由核苷酸序列本身决定;尤其是5′UTR、编码序列(CDS)、3′UTR等关键结构元件,共同影响蛋白表达和药物的体内表现。
近日,中国科学院上海药物研究所李叙潼、郑明月团队在Journal of Advanced Research发表题为Transforming mRNA drug design with AI: From UTR and codon optimization to coordinated design 的综述文章。文章围绕数据基础、评价指标和算法框架,梳理AI在UTR设计、CDS优化和UTR-CDS协同设计中的进展,并指出数据标准化、模型泛化、多目标权衡和可解释性等需要重点解决的问题。引言
mRNA在基因表达和蛋白质合成中承担信息传递作用,已成为疫苗、癌症免疫治疗和蛋白替代疗法的重要技术平台。SARS-CoV-2 mRNA疫苗的大规模临床验证证明,mRNA药物可以在较短周期内完成设计和开发,并在特定适应症中实现有效保护。mRNA药物不需要进入细胞核、不整合基因组,表达具有可逆性和可调控性,且制造流程具有平台化优势。
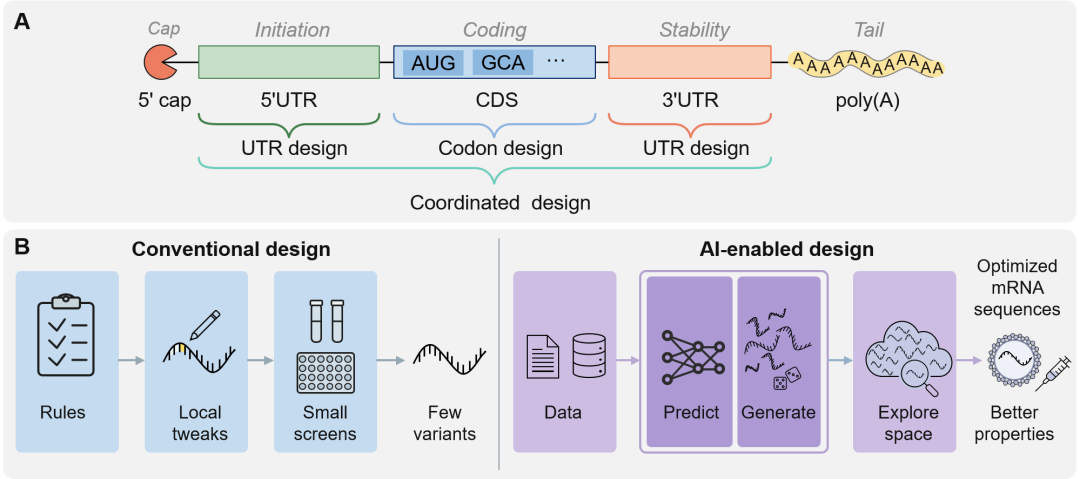
图1. AI在mRNA设计中的总体框架。(A)mRNA结构元件及其优化范围,UTR设计、密码子设计和多区域协同设计分别作用于5′UTR、CDS和3′UTR等调控层级。(B)从传统规则驱动设计向AI驱动设计的转变,AI结合数据预测、生成建模和序列空间搜索,以优先获得性质更优的mRNA候选序列。如图1A所示,从结构上看,成熟真核mRNA由5′端帽结构、5′UTR、编码序列(CDS)、3′UTR和Poly(A)尾等模块构成,并常与假尿苷或N1-甲基假尿苷等核苷修饰共同发挥作用。5′端帽和Kozak序列影响核糖体起始识别,UTR和CDS的二级结构及局部密码子组成共同调控扫描、起始和延伸,3′UTR中的顺式调控元件则参与mRNA定位、稳定性和降解。
从分子设计角度看,mRNA序列优化是在强生物学与生物物理约束下进行的高维组合优化问题。例如,SARS-CoV-2刺突蛋白对应的同义编码空间可达约10632种序列;尽管这些序列编码相同多肽,它们在二级结构稳定性、翻译效率(TE)等关键性质上可能相差数个数量级。如图1B所示,传统策略多依赖启发式规则和低至中通量实验,只能探索局部区域,且难以同时平衡TE、稳定性和免疫原性。此外,将UTR和CDS解耦优化,还可能忽视全长分子内的长程调控作用,降低模型在复杂生物体系中的泛化能力。人工智能(AI)的引入为突破上述组合设计瓶颈提供了新的范式。深度学习表征模型可从大规模异质数据中学习序列、结构与功能之间的非线性关系,用于预测TE、mRNA稳定性、蛋白表达水平及先天免疫激活等关键性质;在此基础上,生成模型和优化模型可将这些预测器作为评分函数,在同义编码序列变体和UTR构型组成的巨大联合空间中进行目标导向搜索,从而实现多种药理学约束之间的更优权衡。随着相关计算模型快速涌现,mRNA设计范式正由单一区域优化转向UTR与CDS的协同设计,包括交替优化、模块化生成、整合评分和全长表征学习等策略。因此,本综述从UTR、CDS及UTR-CDS协同设计三个层次系统梳理AI驱动的mRNA序列工程方法,重点讨论表征模型与生成/优化模型两条方法学主线,并进一步分析数据碎片化、跨体系泛化不足、多目标药理学建模有限及模型可解释性不足等挑战,展望标准化开放数据平台、多模态基础模型和闭环“设计-构建-测试-学习”框架的发展方向。
数据
本综述将AI辅助mRNA设计所依赖的数据概括为多层级体系。第一类数据来自公共数据库和大规模多组学项目,包括Ensembl、RefSeq、GENCODE、UniProt、GTEx和Human Protein Atlas等资源。这些数据库提供CDS和UTR注释、蛋白目标集合、组织表达谱、RNA-seq和Ribo-seq等信息,可用于构建自监督预训练语料、估计密码子使用偏好、分析UTR基序分布以及近似标注翻译效率和核糖体占据情况。公共数据具有覆盖面广的优势,但其多数反映稳态测量而非可控扰动的因果效应;表达水平还受到启动子活性、剪接、核输出和降解等因素共同影响。因此,仅依赖公共数据通常难以精确判断特定UTR片段或少量密码子替换对翻译效率和稳定性的影响。为获得更可控的监督信号,当前多数研究采用合成报告基因文库和大规模并行报告实验(MPRA),在统一载体和启动子背景下系统改变5′UTR、3′UTR或CDS片段,并测定转录输出、核糖体负载、报告活性或蛋白表达。第三类数据来自体外和体内功能验证,其规模较小但更接近药物开发场景,包括不同细胞系中的蛋白表达、抗原表达、mRNA稳定性、组织分布、免疫原性和抗体滴度等终点。综述强调,当前数据景观最突出的问题是高度定制化、原始数据共享不足和统一基准缺乏。不同研究的文库长度、随机化策略、修饰体系和细胞模型差异较大,导致性能指标往往只能在原始数据集内部解释,难以进行严格横向比较。未来需要构建覆盖多细胞类型、多时间尺度和多核苷修饰条件的开放数据集,并配套标准化评测流程。评价指标
mRNA的细胞内生命周期涉及折叠、翻译起始、延伸和降解等耦合过程,因此不存在单一指标能够完整表征药物性质。综述将常用指标分为四类:序列统计和生物物理特征、热力学稳定性、动态翻译动力学,以及药效学和可开发性终点。这些指标既是AI模型的优化目标,也是模型比较和实验验证的基准。结构和序列层面,最小自由能(MFE)常用于描述RNA二级结构稳定性。CDS优化还常使用密码子适应指数(CAI)、最优密码子频率(Fop)和有效密码子数(ENC)等指标,但CAI和Fop依赖宿主背景、参考基因集和计算方法,ENC也只能反映密码子使用偏倚强度,不能直接说明其是否有利于特定宿主翻译。功能层面,平均核糖体负载(MRL)、翻译效率(TE)、mRNA半衰期、蛋白表达水平(EL)和免疫原性更接近药物开发需求。综述指出,临床级mRNA设计必须从单一表达最大化转向多目标优化,在高表达、长半衰期、低免疫原性、组织特异性和制造可行性之间寻找可解释、可验证的折中方案。UTR设计
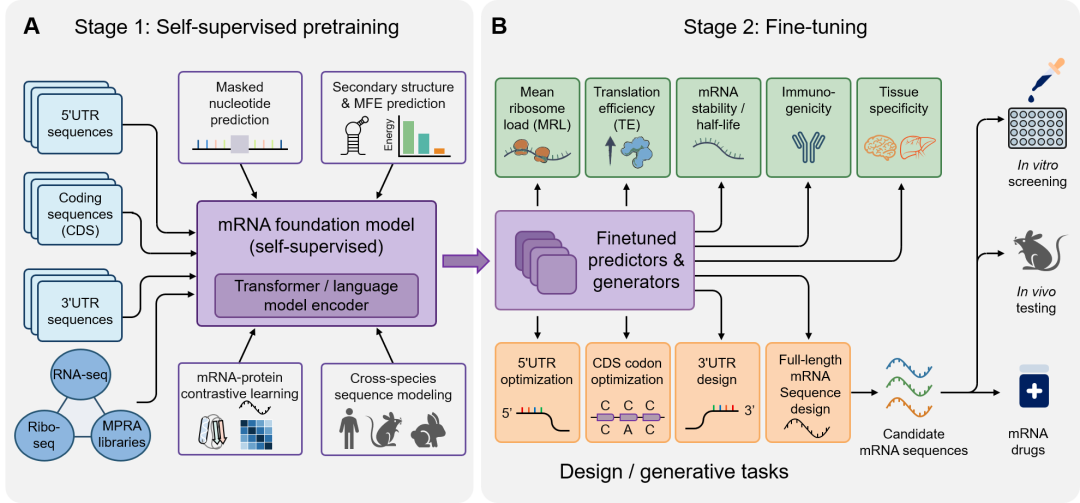
UTR是mRNA药物序列中最直接调控翻译起始、稳定性和细胞类型响应的顺式作用元件。5′UTR影响核糖体识别、扫描和起始密码子选择,3′UTR则通过RNA结合蛋白和miRNA等调控mRNA定位、半衰期和降解。传统UTR设计多借用高表达内源基因UTR模板,或通过Kozak共识序列、删除上游开放阅读框和AU-rich元件等方式降低翻译抑制与降解风险。这些策略有助于早期开发,但仍属于经验迁移,难以系统探索UTR空间。在这一背景下,基于AI的数据驱动方法为突破这些设计瓶颈提供了新机会。对于mRNA治疗药物,AI模型既可以作为预测器,估计候选UTR在指定细胞环境中的翻译水平和稳定性;也可以作为设计器,从头生成满足预设药理属性的合成UTR,从而推动设计范式从经验模仿天然UTR转向精准的性质驱动设计。当前UTR设计正在从单个预测器走向“预训练-微调-实验验证”的基础模型框架。预训练阶段(图2A)不仅可以使用天然5′UTR序列,还可以整合CDS、3′UTR、RNA-seq、Ribo-seq和MPRA文库等多来源数据;模型通过掩码核苷酸预测、二级结构和MFE预测等自监督任务学习序列语法、结构约束和跨物种保守信息。这一步的意义在于,模型不再只记忆某个报告文库中的局部k-mer特征,而是形成对UTR调控语法的通用表征,为后续少量高质量标注数据上的任务微调提供基础。在微调阶段(图2B),预训练模型可以被适配到平均核糖体负载(MRL)、翻译效率(TE)、mRNA稳定性、半衰期、免疫原性和组织特异性等下游任务。同时,预测任务和设计任务可以被放在同一流程中:一方面,模型可作为评分器,对候选UTR预测表达、稳定性或免疫相关风险;另一方面,模型也可作为生成器或优化器,输出5′UTR、3′UTR甚至全长mRNA候选序列。候选序列随后需要经过体外筛选和体内测试,最终才能进入mRNA药物开发场景。图2. mRNA基础模型的预训练与微调框架。(A)利用大规模序列数据库和多组学谱进行自监督预训练,通过掩码核苷酸预测、结构建模等任务学习可泛化表示。(B)预训练模型可在MRL、TE、稳定性、免疫原性、UTR设计和CDS优化等下游任务中微调,并与体外和体内验证衔接。早期UTR建模以判别式预测为主。Optimus 5-Prime利用CNN从MPRA文库学习5′UTR与MRL的关系,并可结合遗传算法进行逆向设计;Frame Pooling加入阅读框信息以适应可变长UTR。随后,Random Forest、MTtrans、UTR-LM和UTR-Insight等方法分别引入手工特征、多任务学习、Transformer预训练和CNN-Transformer解码,推动模型从局部k-mer记忆走向更通用的调控表征。尤其是UTR-LM和UTR-Insight这类模型更接近图2所示框架:先从大规模序列中学习通用表示,再针对MRL、TE或表达水平进行微调和筛选。生成式AI也开始直接参与UTR设计。UTRGAN、Smart5UTR、PARADE和MOBO-5UTR等方法可在潜在空间中搜索高表达、低毒性、组织特异性或多目标平衡的UTR序列;其中Smart5UTR面向m1Ψ修饰mRNA疫苗,PARADE强调细胞类型选择性,MOBO-5UTR则把TE、稳定性、碱基组成和结构风险纳入同一优化框架。总体来看,AI使UTR设计从有限天然模板筛选,迈向可预测、可生成和可多目标优化的新阶段。未来的关键挑战在于提升模型从合成文库到真实内源 mRNA、从体外体系到体内药物场景的泛化能力,并通过实验验证建立更可靠的闭环设计平台。CDS优化
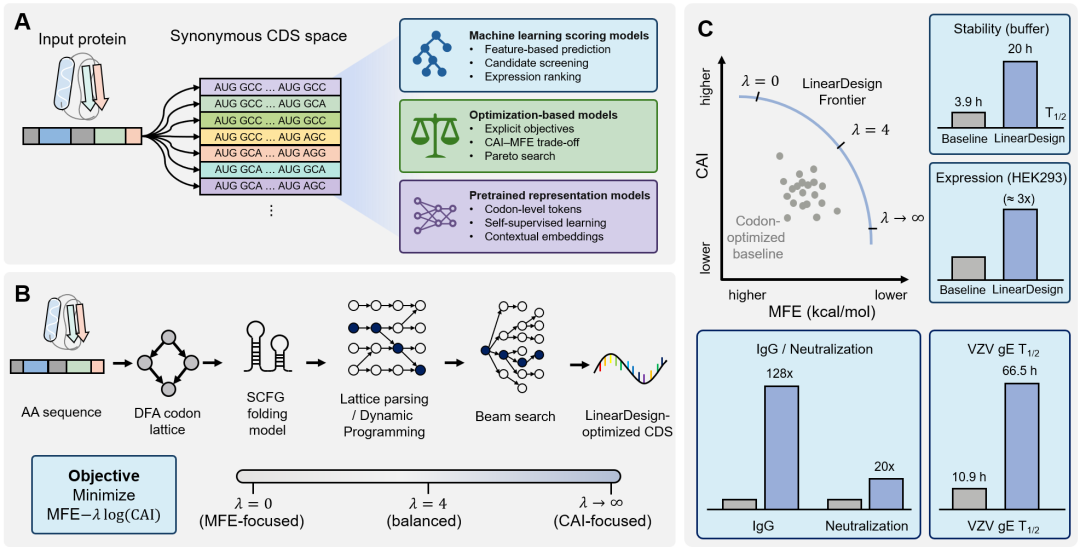
CDS优化是提高mRNA TE、缓解蛋白表达瓶颈的核心策略,其做法是在冗余遗传密码中优先选择有利的同义密码子。由于密码子使用具有简并性,即便是绿色荧光蛋白(GFP)等相对较短的蛋白,也拥有天文数量级的理论同义编码空间;而SARS-CoV-2刺突蛋白等大型病毒抗原则会产生大得多的组合序列空间。这些同义设计空间规模巨大,使穷举枚举和暴力搜索在计算上不可行。高表达宿主通常具有密码子使用偏好,最优密码子也常与更丰富的tRNA池相关,但实际蛋白表达水平由mRNA二级结构、GC含量、稀有密码子簇位置以及翻译起始和延伸动力学共同决定。仅依据CAI等频率指标进行替换,可能耗竭tRNA资源、诱发核糖体停顿,也可能引入不利结构或破坏关键翻译后修饰位点。因此,CDS优化本质上是巨大离散空间中的多目标优化问题。CDS优化模型可以概括为三类(图3A):机器学习评分模型用于预测表达、稳定性或候选排序;优化模型把CAI、MFE和GC含量等指标写入目标函数,在同义CDS空间中搜索折中;预训练表征模型(如CodonBERT)通过自监督学习上下文相关的密码子嵌入,支持表达、降解速率或稳定性预测。三类方法分别提供评分、搜索和表征能力,实际应用中常需组合使用。图3. CDS优化算法及LinearDesign工作流程。(A)给定蛋白或氨基酸序列后,同义CDS空间可通过机器学习评分、优化模型和预训练表征模型等策略探索。(B)LinearDesign通过DFA、SCFG、动态规划和束搜索,在MFE与CAI之间建立可调权衡。(C)不同权重参数可描绘CAI-MFE优化前沿,并用于选择表达和稳定性更平衡的设计。LinearDesign(图3B)是CDS优化的代表算法。它不是深度生成模型,而是基于形式语法和动态规划的计算优化方法:DFA紧凑表示给定蛋白序列的同义密码子空间,SCFG描述RNA折叠能量模型,再通过格解析、动态规划和束搜索寻找候选序列。其目标函数用MFE衡量结构稳定性、用CAI描述密码子适应性,并通过参数λ调节二者权重。如图3C所示,改变λ可以描绘CAI-MFE优化前沿,使研究者可以从“结构更稳定”到“密码子更适应宿主”的连续谱中选择候选序列,而不是依赖单一指标。已有验证显示,优化序列可延长体外半衰期、提高细胞蛋白表达,并增强疫苗相关抗体应答。这些结果说明,CDS优化需要在同一设计空间中协调结构稳定性、翻译效率和免疫效果,而非只追求单一指标最大化。与LinearDesign不同,CodonBERT代表的是预训练表征路线。它以密码子而不是单个核苷酸作为基本token,在大规模多物种CDS上进行自监督训练,从而学习密码子上下文、物种偏好和序列功能之间的潜在关系。这样的模型不直接给出唯一最优CDS,而是为表达预测、降解速率预测、候选序列打分和后续生成模型提供更丰富的表示。综述因此把CDS设计总结为两条互补路线:一类如LinearDesign,明确构造可解释目标函数并搜索优化前沿;另一类如CodonBERT,学习高维序列表征以支持预测和迁移。未来真正实用的CDS优化通常需要把这两者结合,并同时纳入免疫原性、制造性、LNP递送兼容性和跨物种泛化等约束。协同设计
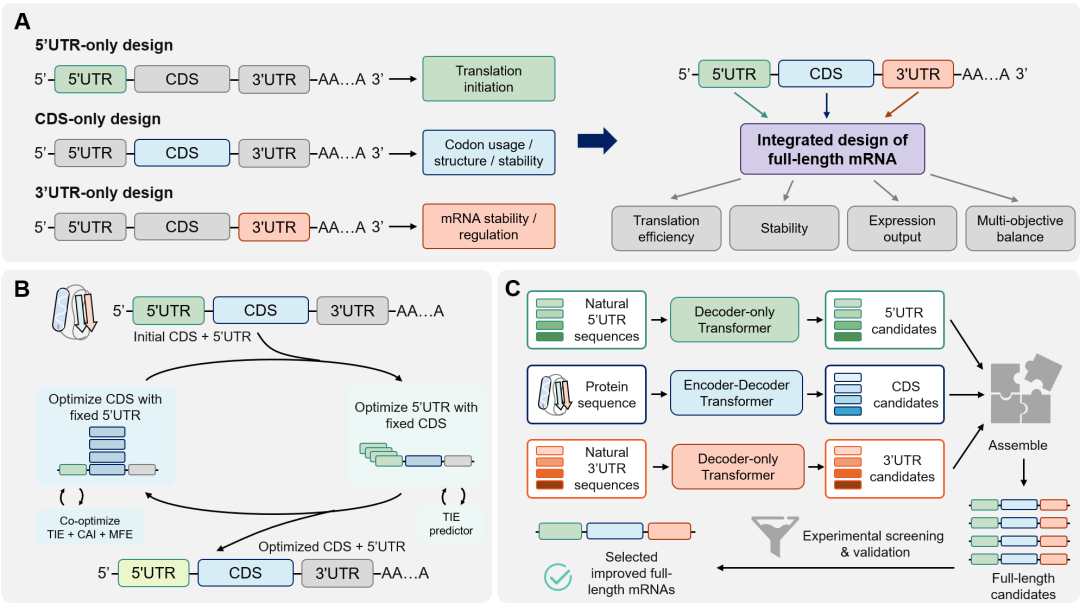
传统mRNA优化常把CDS和UTR视为独立单元,例如只优化CDS的CAI,或只改造5′UTR以增强翻译起始效率(图4A)。这种局部优化可能忽视UTR与CDS之间的结构和功能耦合,难以在多个目标之间达到全局平衡。因此,5′UTR和CDS联合设计成为下一代方法的重要方向,近期模型已开始把UTR-CDS相互作用纳入TE或EL预测,并探索协同优化、模块化生成和下游筛选。图4. 多区域mRNA协同设计策略。(A)从5′UTR、CDS和3′UTR等局部优化走向多区域协同设计。(B)LinearDesign2展示5′UTR-CDS交替优化思路,综合TIE、CAI和MFE等目标。(C)GEMORNA代表模块化全长候选序列生成和筛选框架,体现协同多区域设计,而非完全端到端统一全长模型。LinearDesign2(图4B)是在LinearDesign基础上的5′UTR-CDS交替优化扩展,并非端到端全长生成模型。它加入基于高通量数据训练的TIE预测器,把TIE、CAI和5′UTR-CDS转录本MFE纳入复合目标,在迭代搜索中寻找折中方案。该方法显示了联合优化的价值,但目前仍未显式建模3′UTR调控贡献,且局部邻域搜索对极大设计空间的覆盖能力有限。另一个代表性模型GEMORNA采用模块化生成与筛选框架(图4C)。CDS模块使用Transformer编码器-解码器,根据目标蛋白生成同义CDS;UTR模块采用解码器架构,分别对5′UTR和3′UTR预训练并在高MRL或高稳定性文库上微调。推理时,模型先生成多个CDS候选,再为其搭配不同长度的5′UTR和3′UTR,连接成全长序列后通过少量体外和体内实验筛选。综述强调,GEMORNA应理解为“模块化生成+整合筛选”,而非端到端全长全局模型;当目标和约束增多时,组合空间仍可能带来较重实验负担。mRNABERT则把CodonBERT的表征思想扩展到全长mRNA转录本。它在约1800万条非冗余全长mRNA序列上预训练,显式区分5′UTR、CDS和3′UTR,并采用UTR核苷酸级token、CDS密码子级token的双重token化方案。结合长序列建模和与蛋白语言模型的对比学习,mRNABERT在TE预测、表达和稳定性预测、RBP结合位点识别等任务中表现突出,有望成为协同设计的全长评分骨干;但它目前主要用于表征和判别,还不是实验整合型生成设计框架。从药物开发角度看,协同优化仍处于早期阶段。多数方法的目标集合较窄,对免疫原性、组织/细胞类型特异性、体内药代动力学和可制造性等属性建模不足;跨物种和跨实验语境泛化能力也有限;模型生成或优化的序列往往难以对应到明确调控元件或结构-功能机制。未来框架需要把多维药理学和工程约束纳入统一目标,提高跨体系泛化,并引入更透明的结构分析和归因解释。展望与结论
随着AI与RNA生物学融合,mRNA药物设计正在从经验性试错转向数据驱动的理性设计。设计对象也从UTR局部筛选或孤立CDS密码子适配,拓展到考虑全长转录本长程相互作用和协同优化的模型,使研究者能够在TE、稳定性和免疫原性等多目标空间中更精准地工程化mRNA分子。方法上,AI辅助mRNA设计主要沿表征学习和生成式设计两条路线发展。前者通过自监督预训练或多任务学习建立序列到功能的映射,支持TE和降解动力学等表型预测;后者利用扩散模型、变分自编码器、强化学习或其他搜索策略,在庞大序列空间中寻找满足约束的候选。越来越多的框架开始把高精度表征模型作为评分函数或约束模块,引导生成模型形成闭环优化。文章也指出,现有模型在复杂体内环境中仍有限制:合成MPRA数据与内源转录本、组织环境和病理状态之间存在分布偏移;生成或优化序列难以解释其具体调控机制;同时,LNP递送会受到序列长度、GC含量、结构紧密度和电荷分布等因素影响,而当前模型多将递送体系视为外部变量。面向未来,综述提出四个方向:建立分层、标准化、开放的数据基准;发展整合序列、结构、化学修饰和多组学表型的多模态基础模型;将目标函数扩展到低免疫原性、组织特异性、药代药效、可制造性和LNP相容性等全生命周期属性;建立主动学习和“设计-构建-测试-学习”闭环,用实验反馈持续校正模型偏差。总体而言,AI辅助mRNA设计已显示出加速治疗药物开发的潜力,但从学术模型走向临床应用,仍需解决数据标准化、模型泛化、多目标权衡和可解释性等问题。该综述为理解这一领域的技术格局、关键瓶颈和未来方向提供了系统框架。中国科学院上海药物研究所博士研究生石宇琪、硕士研究生曾传龙为本文共同第一作者;中国科学院上海药物研究所李叙潼副研究员、郑明月研究员为论文共同通讯作者。研究得到上海市科学技术委员会、国家自然科学基金、中国科学院上海药物研究所、中国科学院战略性先导科技专项、上海药物研究所-上海中医药大学中医药创新联合研究项目、上海市科技重大专项和国家重点研发计划等项目资助。Yuqi Shi, Chuanlong Zeng, Xia Sheng, et al. Transforming mRNA drug design with AI: From UTR and codon optimization to coordinated design. Journal of Advanced Research, 2026, Doi: 10.1016/j.jare.2026.06.013.(点击下方阅读原文跳转)
内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢