
DRUGONE
药物所采用的晶型会显著影响其溶解行为、加工性能,甚至临床疗效。然而,对于柔性分子而言,预测其可能形成哪些多晶型,仍然是药物科学中最困难的问题之一。不同晶型之间的能量差通常小于2 kJ mol⁻¹,这种精度虽然可由量子化学方法达到,但计算成本极高,难以用于大规模晶体结构搜索。
研究人员使用AIMNet2机器学习原子间势来探索塞来昔布的多晶型景观。塞来昔布是一种广泛使用的COX-2抑制剂,其晶型I具有创纪录的弹性柔性。研究人员通过基于簇参考数据的主动学习对AIMNet2进行精调,并构建了GPU加速的晶体结构预测流程,用近似量子化学精度生成和排序数十万种候选结构。该流程成功恢复了实验已知晶型I、II和III的能量排序,并以亚埃级几何精度重现实验晶体结构,同时识别出两个距离最稳定已知晶型能量低于4 kJ mol⁻¹的低能候选结构。
混合密度泛函理论计算也给出了类似的浅能量景观,显示多个多晶型在热力学上处于竞争状态。有限温度分析进一步揭示,对于晶型I这类超柔软晶体,静态晶格模型存在明显局限。研究人员认为,该框架不仅为塞来昔布实验多晶型筛选提供了有物理依据的候选目标,也为柔性药物分子的晶体结构预测提供了一种可迁移策略。

多晶型是指同一化合物能够以不同晶体结构结晶的现象,在有机固体中非常常见。对于活性药物成分而言,不同晶型可能表现出不同的溶解度、溶出速率、生物利用度、力学性质、可加工性和专利价值。因此,在药物开发和制剂研究中,发现、表征和控制多晶型是非常关键的环节。
塞来昔布是一种非甾体抗炎药,也是首个获批用于类风湿关节炎和骨关节炎治疗的选择性COX-2抑制剂。由于其临床重要性和较差水溶性,塞来昔布的多晶型长期受到关注。目前已报道四种晶型,即晶型I至晶型IV。其中,晶型III被认为是在环境条件下热力学稳定的晶型。晶型III的单晶结构较早被解析,而晶型II的结构直到多年后才得到报道。晶型I由于难以获得适合单晶衍射的晶体,其结构解析更晚完成;最新单晶结果显示,晶型I具有超过8.7%的弹性应变,刷新了有机分子晶体的弹性柔性记录。晶型IV则仍然缺乏明确结构表征。
这些现象表明,塞来昔布具有非常浅的自由能景观,多个晶型可能具有接近的能量。这也意味着,还可能存在尚未被实验表征的晶型。如果这些潜在晶型具有更好的溶解性、力学性能或加工性质,它们就可能在药物开发中具有实际价值。因此,系统开展多晶型筛选和计算预测非常重要。
计算晶体结构预测已经成为药物多晶型研究的重要工具。传统方法通常依赖经验力场或密度泛函理论。经验力场计算速度较快,但精度有限;高精度周期性DFT方法更可靠,但计算成本极高,尤其不适合大规模筛选柔性分子。塞来昔布这类体系尤其困难,因为它具有较高分子柔性,且某些晶型中不对称单元内包含多个独立分子。候选晶型之间的能量差又很小,因此任何微小计算误差都可能改变晶型排序。
机器学习原子间势为这一问题提供了新机会。它们可以以远低于量子化学的成本,逼近DFT能量和力,从而支持大规模晶体结构生成、优化和排序。研究人员因此使用AIMNet2机器学习原子间势,并通过主动学习和量子化学簇数据进行精调,以系统探索塞来昔布的多晶型景观。
方法
研究人员构建了一个基于AIMNet2的晶体结构预测流程。该流程从塞来昔布二维分子结构出发,首先生成合理的三维构象,然后根据空间群、晶胞中分子数和不对称单元内独立分子数构建大量候选晶体结构。为实现大规模采样,研究人员开发了基于PyTorch和GPU加速的晶体结构生成器。随后,研究人员使用主动学习策略精调AIMNet2模型:先用初始AIMNet2优化候选晶体结构,再从优化后的晶体中提取代表性分子簇,对这些簇进行量子化学计算以获得参考数据,并用这些数据继续训练AIMNet2。经过迭代后,精调模型能够快速评估大量候选晶体的相对晶格能,并用于预测0 K能量景观。研究人员进一步使用声子计算,在谐近似和准谐近似下分析温度依赖的亥姆霍兹自由能,同时计算不同晶型的杨氏模量,以评估其稳定性、加工相关性质和力学表现。
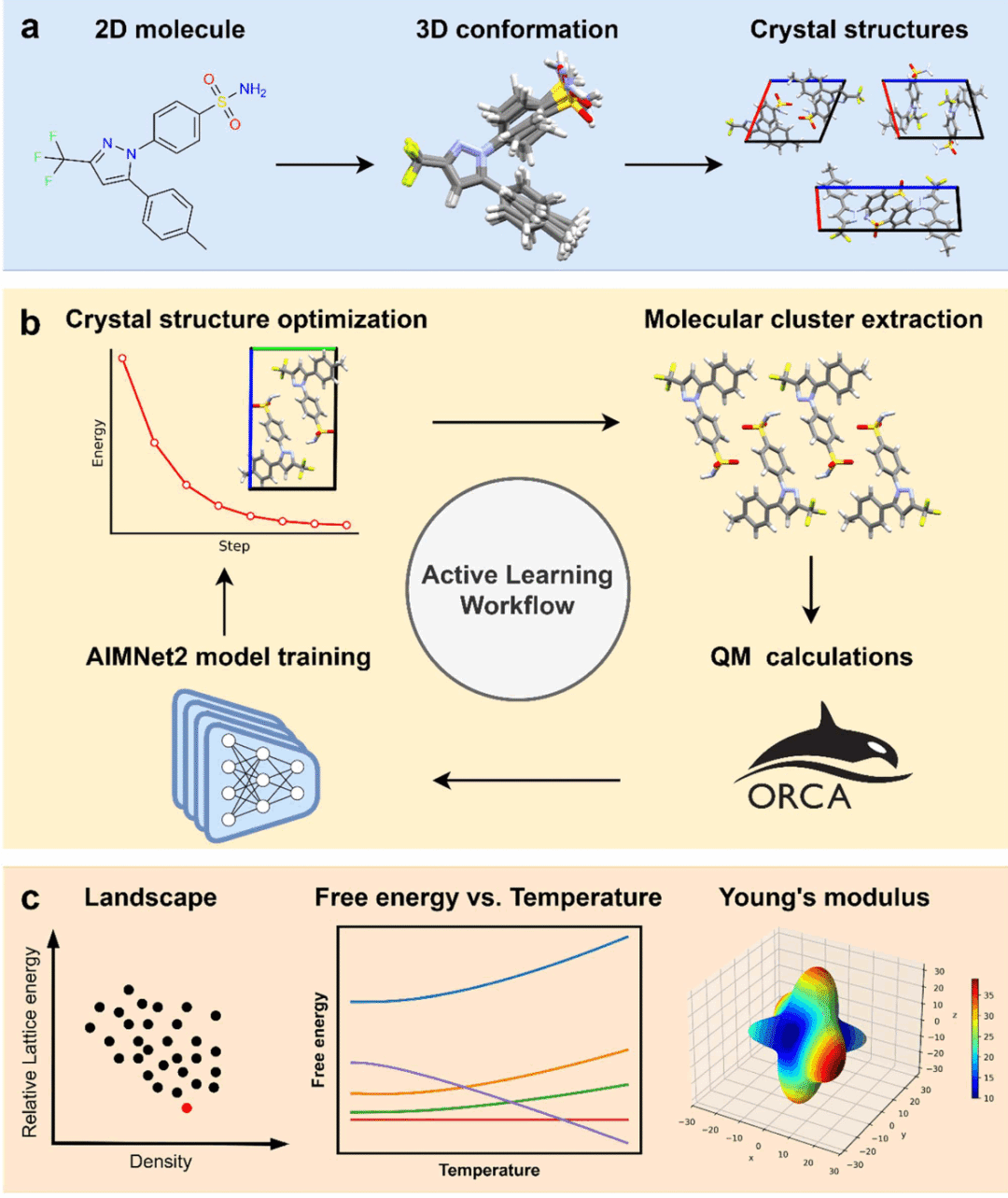
图1|塞来昔布晶体结构预测工作流概览。
结果
基于AIMNet2的分子晶体预测工作流
研究人员首先建立并验证了AIMNet2晶体结构预测流程。该流程包括晶体结构生成、主动学习精调和性质预测三个核心部分。与传统晶体结构预测方法相比,该流程的一个重要优势是能够在保留全柔性晶体结构优化的同时,处理数以万计甚至更多候选结构,而不会过早丢弃可能具有物理意义的低能候选晶型。
主动学习阶段虽然需要对超过两万个分子簇进行量子化学计算,但这些计算是在分子簇而非完整周期晶体上完成的,因此成本远低于对大量候选晶体执行周期性DFT计算。精调后的AIMNet2能够较好重现PBE + MBD相对晶格能,平均绝对误差约为1.84 kJ mol⁻¹。考虑到药物多晶型之间的能量差通常只有1–2 kJ mol⁻¹,这一精度足以用于大规模初筛和初步排序,尤其适合在昂贵周期性DFT计算之前缩小候选范围。
研究人员强调,该流程并没有完全取代DFT,而是将计算成本重新分配到一个接近量子化学精度的神经网络势能模型上。这样既减少了昂贵周期性DFT计算的数量,又能更完整地探索晶体能量景观。
多晶型能量景观
研究人员从三个角度评估精调AIMNet2模型:是否能在候选结构景观中识别已知晶型,是否能准确给出相对能量排序,以及优化后的几何结构是否与实验晶体结构一致。
在0 K相对晶格能景观中,AIMNet2能够将已知的晶型II和晶型III定位在低能区域,说明模型具有较好的多晶型识别能力。晶型I的晶格能明显高于晶型III,约高8.86 kJ mol⁻¹,这与实验上晶型III更稳定的认识一致。AIMNet2预测晶型II比晶型III仅高0.12 kJ mol⁻¹,这一差值小于模型本身的能量不确定性,因此更合理的解释是:模型认为晶型II和晶型III在能量上非常接近,而不是对二者绝对排序作出强判断。
为了进一步检验能量排序的稳健性,研究人员还使用混合密度泛函方法重新计算了五个选定晶型的相对晶格能。尽管不同泛函和优化几何会导致绝对能量差有所不同,但所有方法都一致显示:塞来昔布具有浅能量景观,多个结构处于数kJ mol⁻¹范围内的低能竞争状态。特别是候选结构#1、晶型II和晶型III在多种方法下始终位于低能区域,候选结构#2也通常位于全局最低能结构附近。
温度依赖自由能分析进一步显示,当加入零点能和热修正后,两个此前未报道的低能晶体结构候选物#1和#2在300 K下也具有竞争力。其中,#1和#2的自由能被预测为低于晶型III,且密度明显更低。这提示可能存在尚未发现的稳定或亚稳定晶型。不过,研究人员也强调,这些结构目前只是基于热力学计算的预测,并未被实验观察到。低晶格能或低自由能并不保证晶体一定可以在实验中获得,因为成核动力学、溶剂效应、结晶路径和晶体生长可及性都可能阻止某些热力学竞争结构形成。
尽管如此,#1和#2与已知稳定晶型之间的自由能差很小,处于实验多晶型筛选通常关注的能量窗口内。因此,它们更适合作为未来实验筛选的有物理依据候选目标,而不是被直接宣称为新的已发现晶型。
在几何结构方面,AIMNet2优化后的晶型I、II和III与实验晶体结构高度一致。三个已知晶型的20分子簇RMSD分别为0.115 Å、0.231 Å和0.168 Å,均低于晶体结构预测基准中通常认为较好的0.3 Å阈值。这说明AIMNet2不仅能进行能量筛选,也能较准确地保持晶体堆积几何。
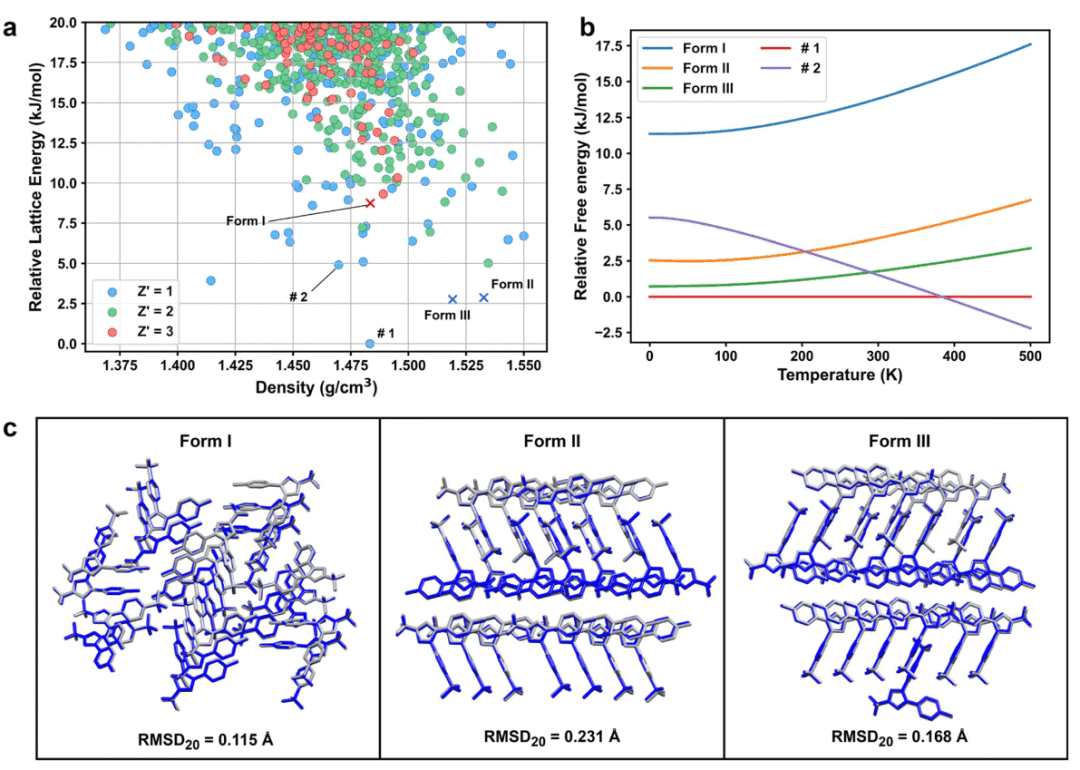
图2|塞来昔布多晶型能量景观、温度依赖自由能和结构重叠。
弹性性质
研究人员进一步计算了塞来昔布不同晶型的杨氏模量,以评估模型对晶体力学性质的预测能力。晶型I尤其重要,因为它具有异常高的弹性柔性,是有机分子晶体中的极端案例。
在0 K静态晶格优化条件下,无论是PBE + MBD还是AIMNet2,都明显高估了晶型I和晶型II的杨氏模量。对于晶型I,实验杨氏模量约为3.18 GPa,而PBE + MBD和AIMNet2分别预测约20.7 GPa和19.1 GPa。对于晶型II,实验值约为16.27 GPa,PBE + MBD和AIMNet2分别预测约22.7 GPa和24.6 GPa。
研究人员认为,这并不意味着实验结果不可靠。相反,晶型II在一些理论处理下与实验符合较好,说明实验测量本身具有可信度。晶型I的巨大差异更可能反映出静态晶格模型的物理局限。0 K静态晶格计算忽略了热膨胀和强非谐效应,而晶型I作为超柔软晶体,其室温有效刚度显然受到这些因素显著影响。准谐近似只能部分捕捉这种软化,因此需要更明确的非谐方法才能准确描述晶型I的宏观弹性响应。
为检验热膨胀影响,研究人员还在固定实验晶胞参数的条件下优化分子几何并计算杨氏模量。AIMNet2显示,随着温度相关晶胞膨胀被纳入,三种晶型的刚度均明显降低。对于晶型II,AIMNet2预测值约为16.1 GPa,与实验值非常接近。相比之下,晶型I仍然无法被准确再现实验中的超低刚度。这表明,晶型I可作为分子晶体有限温度非谐弹性方法的严格基准。
热膨胀对多晶型稳定性的影响
研究人员使用AIMNet2在300 K下对五个选定晶型进行了准谐近似计算,以考虑热膨胀对多晶型稳定性的影响。结果显示,不加入热修正时,AIMNet2认为晶型II和晶型III非常接近;PBE + MBD则倾向于认为晶型II更稳定,并且与候选结构#1自由能相近。混合DFT计算也支持类似结论,即晶型II、晶型III、候选结构#1和#2都处在一个狭窄的低能集合中。
当加入谐近似热修正后,晶型II与晶型III之间的自由能差增加,晶型III与候选结构#2的稳定性变得更加接近。进一步加入准谐近似热膨胀后,AIMNet2预测的整体排序变化不大,只在晶型III和候选结构#2之间出现轻微重排,而二者自由能差仅约0.23 kJ mol⁻¹。这样的差距极小,说明在热力学上它们几乎可以视为竞争晶型。
研究人员还比较了五个晶型的晶体堆积模式。候选结构#1和#2都表现出分子间双齿氢键,而晶型II和晶型III的氢键模式非常相似。这种相似性可能解释了实验中晶型III样品中经常出现晶型II杂质的现象。分子构象比较显示,不同Z′ = 1晶型之间的主要差别来自吡唑环相对于磺酰苯基和对甲苯基的扭转角,以及磺酰胺–NH₂基团绕S–N键的取向差异。
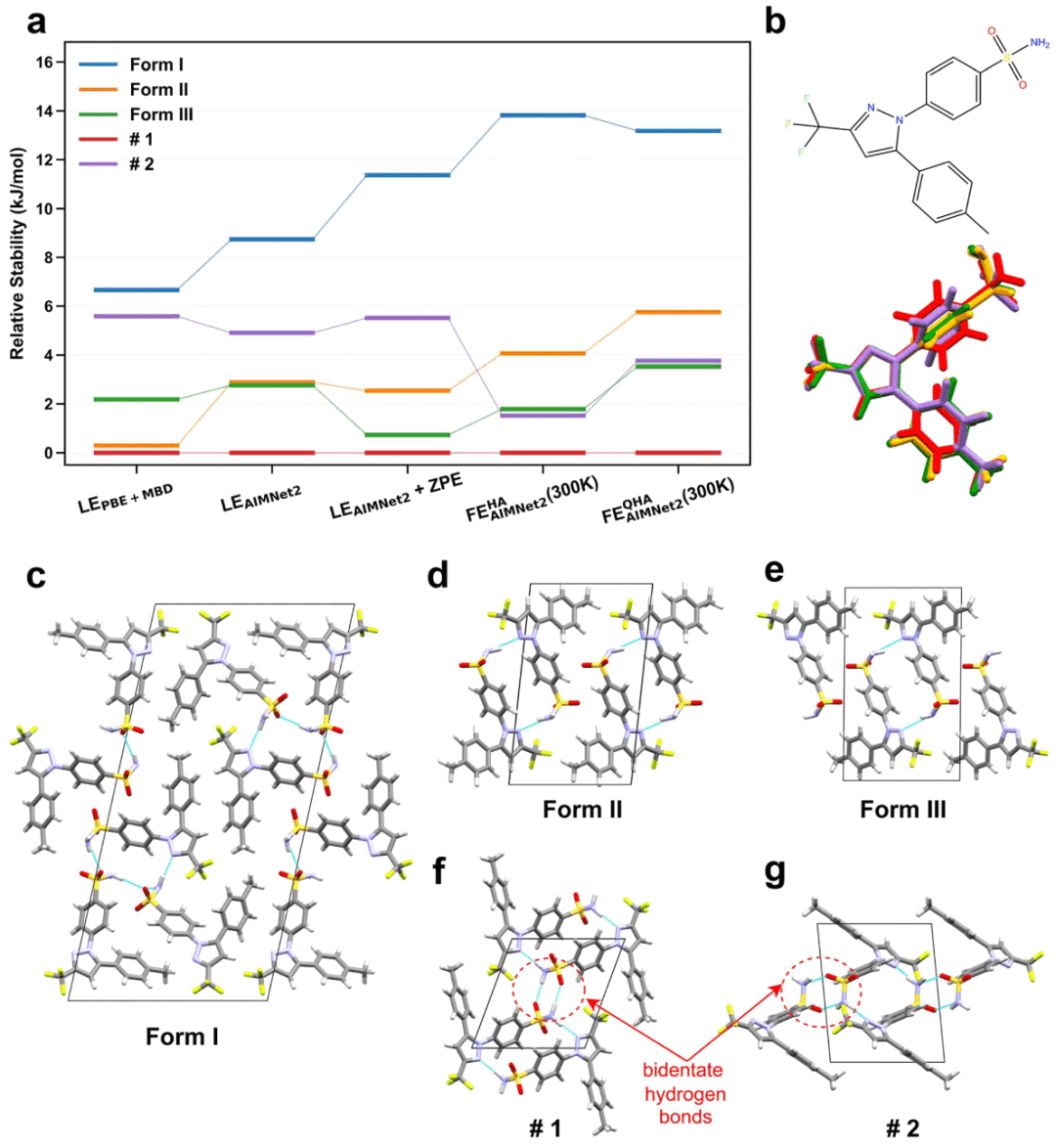
图3|五个塞来昔布候选晶型的相对稳定性、分子构象和晶体堆积。
讨论
研究人员开发了一个由AIMNet2机器学习原子间势驱动的完整晶体结构预测流程,用于研究塞来昔布的多晶型行为。该流程结合GPU加速晶体结构生成、主动学习精调、能量排序、有限温度自由能分析和力学性质预测,成功重现了已知晶型的实验稳定性层级,并预测出两个尚未实验报道的低能候选结构。
这一结果说明,经过簇主动学习精调的AIMNet2能够以远低于周期性DFT的成本,接近量子化学精度地探索柔性药物分子的晶体能量景观。对于塞来昔布,AIMNet2不仅能恢复晶型I、II和III的结构几何,还能在低能区域识别多个热力学竞争晶型。候选结构#1和#2与已知稳定晶型III的自由能差小于4 kJ mol⁻¹,处于药物多晶型研究中通常认为值得关注的范围内。它们具有不同的堆积方式和氢键模式,因此可作为未来实验多晶型筛选的具体结构假设。
不过,研究人员非常谨慎地指出,计算预测不能直接等同于实验发现。一个晶型即使在自由能上较低,也可能因成核路径、结晶动力学、溶剂环境或生长条件限制而无法被实验分离。因此,#1和#2应被理解为有物理依据的实验筛选目标,而不是已经确认的新晶型。未来可以通过溶剂介导转化、熔融结晶或其他结晶条件探索来验证这些候选结构是否可及。
本研究也揭示了当前方法的局限。尤其是晶型I的杨氏模量预测问题表明,静态晶格MLIP计算和准谐近似仍无法捕捉超柔软有机晶体中的强非谐弹性响应。对于这类体系,未来需要引入自洽声子、分子动力学弹性常数或其他显式非谐方法,以更准确地描述有限温度下的力学性质。
总体来看,研究人员建立的簇主动学习AIMNet2流程具有较强可迁移性,可用于其他柔性活性药物成分的晶体结构预测。它能够在早期药物开发阶段更快地绘制多晶型景观,帮助研究人员识别潜在稳定晶型、理解晶体堆积差异,并为实验筛选提供更有针对性的结构目标。
整理 | DrugOne团队
参考资料
Zheng, Peikun, Yuriy Abramov, Changquan Calvin Sun, and Olexandr Isayev. "Exploring Celecoxib Polymorph Landscape Using AIMNet2 Machine Learning Interatomic Potential." Chemical Science (2026).

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢