
RNA 可变剪接是转录后调控体系中的核心环节。同一条前体 mRNA 可以通过不同外显子组合、不同剪接位点选择和内含子保留等方式生成多种 RNA 异构体,进而拓展蛋白质功能多样性,并影响 mRNA 稳定性、定位、翻译效率和细胞命运调控。超过 90% 的人类基因会发生可变剪接,这也使其成为理解基因表达调控、疾病发生机制和精准医学应用的重要入口。
问题在于,剪接调控并不是由单一局部序列信号决定的。5′ 和 3′ 剪接位点、分支点、聚嘧啶区、剪接增强子和沉默子、RNA 结合蛋白、组织特异性环境以及远端顺式调控元件,共同塑造最终的剪接结局。因此,如何从基因组序列和多组学数据中准确预测剪接位点、剪接模式以及突变对剪接的影响,已经成为基因组学与精准医学共同面对的关键问题。
近年来,短读长 RNA-seq、长读长转录组、单细胞与空间转录组学持续提高了剪接观测的分辨率;与此同时,深度学习和基因组基础模型快速发展,使研究者开始尝试从更长序列、更复杂上下文和更精细组织背景中学习剪接调控规律。AI 驱动的剪接预测,也正在从早期的剪接位点识别,走向突变效应解释、组织特异性预测和治疗策略设计。

2026年6月25日,浙江大学良渚实验室沈宁课题组在Nature Genetics 发表了综述论文 “Advances and challenges of splicing prediction with AI” ,系统梳理了AI驱动的 RNA 剪接预测算法从统计模型、简单机器学习到深度学习、基因组大模型的发展脉络,重点讨论了模型设计、剪接事件定量、突变效应预测等技术细节,并总结了剪接预测算法的临床转化应用及未来的挑战。
和许多从生物学机制或疾病应用出发的综述不同,这篇文章的特色是从“算法如何被设计出来”这一技术角度切入,不单单介绍“有哪些工具可用”,也进一步讨论这些工具为何这样设计、不同设计的长处与短处。文章围绕训练数据规模、预测输出分辨率、剪接事件定量方式和模型复杂度等核心维度,比较了不同算法在建模逻辑和应用场景上的差异。
整体来看,该综述表明,可变剪接预测正在经历从局部规则到长序列建模、从单一剪接位点到多层级输出、从通用预测到组织和疾病背景解释的连续升级。随着模型能力增强,剪接预测算法也正在被用于临床变异注释、疾病机制解析、剪接来源新抗原发现以及 ASO、小分子和 CRISPR 等剪接靶向治疗设计。
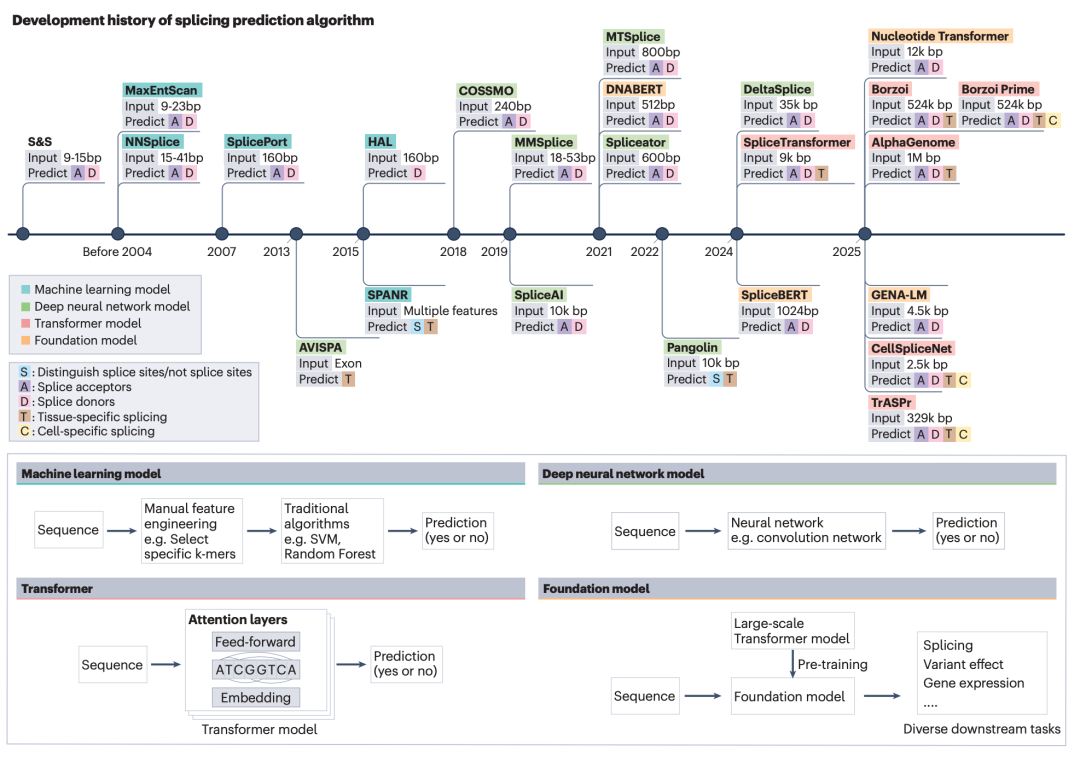
图1: 可变剪接预测算法的演进过程。
文章首先回顾了可变剪接预测算法的历史演进。早期方法主要围绕剪接位点附近的短序列窗口建立统计规则,例如 Shapiro-Senapathy 方法通过位置权重矩阵描述剪接位点碱基频率,MaxEntScan 则进一步利用最大熵模型捕捉相邻核苷酸之间的依赖关系。这一阶段的模型更像是在剪接位点附近寻找固定的“信号词”,优势是计算高效、解释直观,而劣势是难以处理远端调控和复杂上下文。
进入机器学习和深度学习阶段后,模型开始从人工特征设计转向自动特征学习。NNSplice 等早期神经网络方法为剪接位点预测提供了机器学习框架,而 SpliceAI、Pangolin 等基于卷积神经网络和残差结构的模型,则通过读取更长序列上下文显著提升了剪接位点和突变效应预测能力。随着人类对生物机制理解的加深,以及计算技术的进步,研究者们在设计预测算法时,都越来越注重模型计算远端序列特征的能力。此后的Transformer 架构和基因组语言大模型进一步扩展了模型对长距离依赖关系的捕捉能力,使 SpTransformer、SpliceBERT、Nucleotide Transformer、Borzoi 和 AlphaGenome 等模型能够处理组织特异性、长序列调控和多任务预测问题。
从文章总结的演进路线可以看出,可变剪接预测的核心问题已经从“能否识别剪接位点”逐步推进到“能否理解不同序列背景、组织背景和突变背景下的剪接调控逻辑”。模型读取的序列范围越来越长,输出任务越来越多样。于此同时,经由文章总结的表格可以看出,算法的训练数据和算力需求也在高速增长。
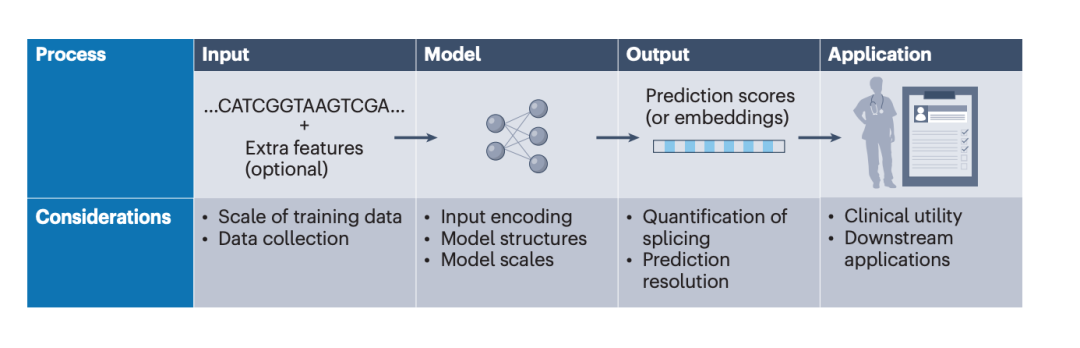
图2: 剪接预测模型的设计框架。
在算法名称之外,本文更重要的贡献是拆解不同剪接预测模型的底层设计差异。作者将模型开发过程概括为输入数据、序列表征与模型结构、预测输出和下游应用等相互连接的阶段。不同算法之间的差异,往往并不只来自模型名称,而是来自训练数据来源、输入序列长度、编码方式、网络结构、预测分辨率和应用目标的组合选择。
训练数据规模是推动模型能力提升的重要因素。早期模型依赖有限的人工整理剪接位点或 cDNA 数据,而现代深度学习模型则吸纳了越来越多的数据,包括不同实验来源(RNA-seq、MPRA)、不同物种,的数据。为了解决有监督数据不足的问题,最近的基因组大模型甚至会使用大规模未标注序列进行无监督预训练。数据规模扩大使模型能够学习更多剪接模式,也带来了数据质量、组织覆盖度、标签定义和计算成本等新问题。
模型结构同样决定了算法能够捕捉何种生物学信息。传统机器学习依赖人工特征,卷积神经网络适合识别局部序列 motif,Transformer 和自注意力机制则更擅长建模远距离依赖关系。近年来的基础模型通常通过大规模预训练获得通用序列表征,再迁移到剪接预测、变异效应评估和基因表达预测等下游任务中。
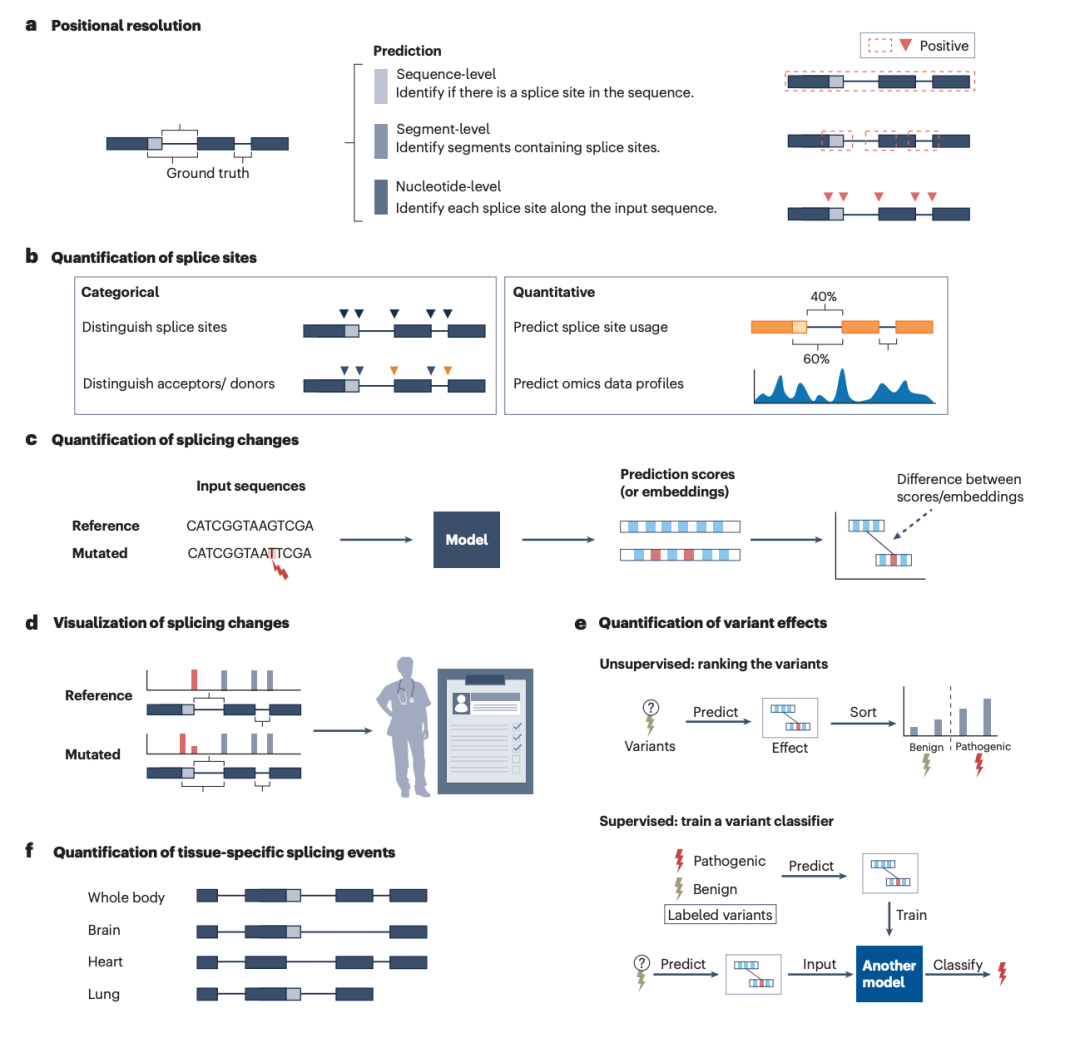
图3: 剪接预测输出分辨率与剪接事件定量。
鱼与熊掌不可兼得,基于训练数据、算力条件等方面的取舍,模型最终表现出的工作方式也各有千秋。文章从多个角度分析了不同算法输出数据的差异:
首先,文章比较了不同模型的预测输出分辨率。序列级模型通常对一段输入序列给出一个整体分数,适合判断某一窗口是否包含剪接信号;片段级模型将序列划分为更小片段并输出对应预测,能够兼顾较长上下文和一定位置分辨率;核苷酸级模型则为每个碱基赋予剪接供体或受体概率,更适合精确定位剪接位点和可视化突变造成的局部变化。
其次,除了预测“哪里是剪接位点”,现代模型还需要回答“剪接事件发生到什么程度”。因此,不同算法采用了多种剪接定量方式,包括二分类或多分类标签、外显子包含比例 PSI、剪接位点使用强度、RNA-seq coverage 以及组织特异性评分等。定量方式直接影响模型的生物学解释力:有些模型更适合做位点识别,有些模型更适合估计外显子纳入水平,有些则试图从测序覆盖度中推断更复杂的转录本结构。
此外,突变效应预测是当前剪接算法最重要的应用之一。多数工具通过比较参考序列和突变序列的模型输出,计算 delta score 或类似指标,从而评估突变是否可能创造新的剪接位点、破坏原有剪接位点或改变剪接强度。核苷酸级模型还能将突变前后的预测曲线直接可视化,使研究者观察隐匿剪接位点激活或外显子边界移动等具体变化。
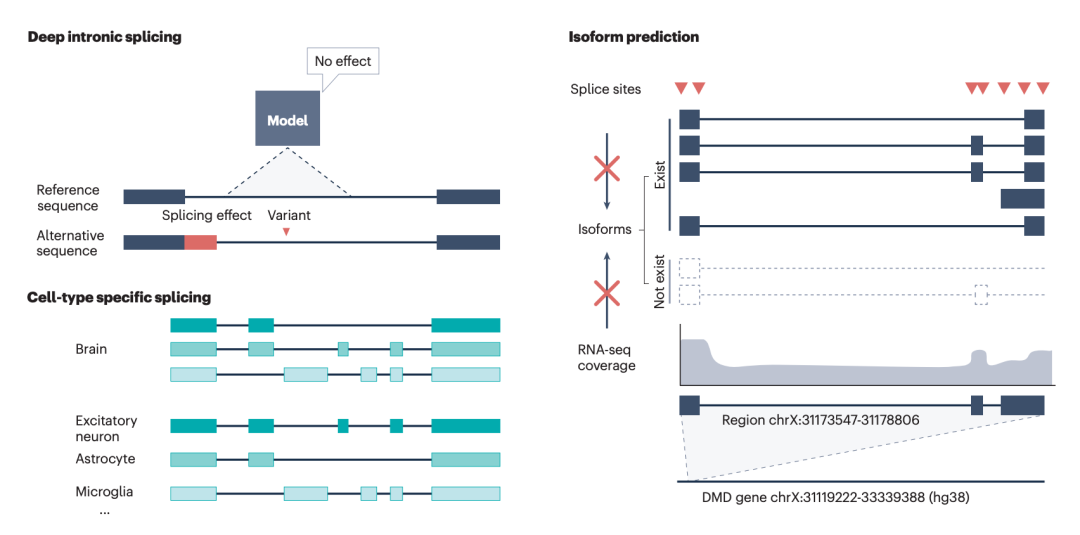
图4: 剪接预测算法面临的关键挑战。
文章强调,尽管深度学习显著提升了剪接预测能力,但当前模型仍面临一系列难题。比如,深内含子变异预测仍不充分。许多致病变异位于远离经典外显子边界的位置,可能通过激活隐匿剪接位点或扰动远端调控元件改变剪接,但相关训练数据相对稀缺,现有模型在远距离突变上的性能仍会下降。
其次,异构体水平重建仍是瓶颈。许多模型能够较好地预测供体位点和受体位点,却难以准确连接多个外显子并恢复完整转录本结构。对于包含多个可变剪接事件的基因而言,仅知道单个剪接位点是否变化,并不足以解释最终异构体组成及其功能后果。
另外,组织和细胞类型特异性预测仍依赖大规模高质量数据。Pangolin、SpTransformer 和相关模型已经开始引入多组织或细胞类型信息,但剪接调控具有高度上下文依赖性,不同组织、细胞状态和疾病环境下的剪接模式可能显著不同。未来单细胞、空间转录组和长读长数据的积累,将是推动模型进一步发展的关键。
讨论
剪接预测的临床转化价值
计算剪接预测已经成为临床基因组学和疾病研究中的重要辅助工具。大量疾病相关变异可能通过扰动剪接发挥作用,尤其是深内含子变异、非编码变异和隐匿剪接位点相关变异,往往难以仅凭常规注释直接解释。SpliceAI、MaxEntScan、SpTransformer、CADD-Splice、MAGPIE 等工具正在被用于变异优先级排序、致病性证据补充和组织特异性剪接异常识别。需要注意的是,剪接预测结果本身并不等同于临床诊断结论。文章强调,模型分数应作为支持性计算证据,与 RNA 实验、功能验证、遗传共分离、临床表型和既有数据库证据等多维度信息结合,共同解释突变影响。也正因如此,未来剪接预测模型的重要方向并不仅是提高分数准确性,还包括提升可解释性、可验证性和与临床证据框架的兼容性。
从剪接改变到治疗设计
除了变异注释,剪接预测还正在进入治疗设计环节。剪接异常可通过反义寡核苷酸、siRNA、小分子和 CRISPR 等方式进行干预,其中 ASO 是目前最成熟的方向之一。AI 模型可辅助识别异常剪接事件、筛选候选靶点、预测寡核苷酸序列效果,并为剪接纠正策略提供更高通量的设计依据。
在肿瘤研究中,异常剪接还可能产生肿瘤特异性新抗原,拓展免疫治疗候选靶点范围。将剪接预测与 RNA-seq、免疫肽组学和变异注释结合,有望发现传统 SNV 和 INDEL 流程之外的肿瘤特异性表位,为个体化癌症疫苗和免疫治疗提供新的候选来源。
大模型并不自动等于更好的剪接预测
随着基因组基础模型兴起,模型参数规模和输入序列长度快速增长。Nucleotide Transformer、Borzoi、AlphaGenome 等模型显示了长序列、多任务和预训练框架的潜力,但文章也指出,模型越大并不必然意味着在所有剪接任务上表现更好。剪接预测的成功取决于生物学任务定义、训练标签质量、组织背景覆盖度、模型结构和计算成本之间的平衡。对于某些明确任务,小型、任务特异性模型可能仍然更高效、更易解释,也更适合实际部署。未来领域需要的不只是更大的模型,还需要能将 DNA/RNA 序列、染色质状态、RNA 结合蛋白占位、RNA 结构、表观转录组修饰和蛋白质组信息整合起来的多模态框架,从而同时捕捉顺式元件和反式调控背景。
值得一提的是,围绕“基因组大模型是否一定优于任务特异性模型”这一问题,沈宁课题组近期也在 Nature Communications 发表了题为“Benchmarking pre-trained genomic language models for RNA sequence-related predictive applications”的相关研究。该研究系统评估了 11 个预训练基因组大模型在多类 RNA 序列相关任务中的表现,在可变剪接位点预测的评估中,文章主要对比了预训练大模型与SpliceAI、SpliceTransformer等小型模型的表现差异。结果表明,预训练大模型在训练数据有限或类别高度不平衡的场景中具有明显潜力,但模型规模并不自动转化为更优性能;预训练数据、输入上下文长度、tokenization 策略、微调方式以及具体生物学任务本身,都会共同决定模型表现。这一结论也与本文综述中对剪接预测模型复杂度的讨论相互呼应:未来 RNA AI 模型的发展,不只是继续扩大参数规模,而是要在数据、算法与生物学问题之间建立更合适的匹配关系。
总结
总体而言,可变剪接预测已经从早期统计规则和短序列打分,发展到深度学习、Transformer 和基因组基础模型驱动的长序列建模阶段。过去,算法主要回答“哪里可能是剪接位点”;现在,模型正在进一步尝试回答“突变如何改变剪接、这种改变在哪些组织中发生、是否可能导致疾病,以及能否被治疗干预”。在可变剪接问题上,AI未来能做的,很可能比现在已经做到的更多。
对于有兴趣设计和开发RNA相关算法的研究者来说,这篇综述在计算模型设计角度有独特的价值。它表明,未来的关键不只是一味构建更大的模型,而是能否把训练数据、模型结构、剪接定量、异构体重建、组织特异性和临床证据整合为同一套可预测、可解释、可验证的框架,并最终服务于精准诊断和剪接靶向治疗。
浙江大学良渚实验室沈宁研究员为该论文的第一作者暨通讯作者。团队成员刘畅和游宁远对该工作作出了重要贡献。
良渚实验室沈宁课题组围绕“组学与精准医学分析算法开发与应用”开展临床转化密切相关的研究,运用生物信息学数据整合分析与人工智能算法,并结合实验筛选平台进行药物研发与精准治疗。课题组目前有多项具有重要应用价值的课题正在推进,与医学专家主导的实验室保持合作,诚招具有实验生物、计算生物背景的博士后和研究助理。
详细招聘信息见:https://person.zju.edu.cn/shenning
简历投递:shenningzju@zju.edu.cn
参考资料
Shen, N., You, N. & Liu, C. Advances and challenges of splicing prediction with AI. Nat Genet (2026).
https://doi.org/10.1038/s41588-026-02629-4
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢