

文献来源:Persico, M.; Micoli, A.; Salmaso, V.; Cianciulli, A.; Moro, S.; Spalluto, G.; Buccioni, M.; Marucci, G.; Volpini, R.; Pozzan, A.; Micheli, F.; Federico, S. Applying Deep-Learning-Driven De Novo Design to Hit Identification: A Case Study on A2A Adenosine Receptor Antagonists.J. Med. Chem. 2026.
DOI: 10.1021/acs.jmedchem.6c00231
通讯作者单位:Aptuit(Evotec子公司,意大利维罗纳);的里雅斯特大学化学与药学系;帕多瓦大学分子建模研究组(MMS)
导读
本文是一篇方法学透明度较高的AI从头药物设计实证研究。研究团队使用 AstraZeneca 开发、Evotec 定制化改造的生成式工具 REINVENT 3.2,以 A2A 腺苷受体(A2A AR) 为模型靶点,构建了融合"配体活性预测—对接—血脑屏障渗透性"三组分的多参数优化(MPO)评分函数,驱动强化学习循环生成新分子。
全文围绕两个核心变量展开系统性的对照实验:
1. 评分函数的参数化方式(各组分权重、是否引入结构约束惩罚项); 2. 生成后过滤管线的设计逻辑(过滤顺序、过滤类型、是否引入机器学习排序方法)。
在保持生成阶段产出的化学空间基本不变的前提下,仅通过调整过滤策略与评分函数权重,实验命中率从 0% 提升到 100%,活性从"完全无效"提升到"高纳摩尔级"。这一结果为AI驱动的从头设计提供了一个具有方法学参考价值的失败-修正完整案例。
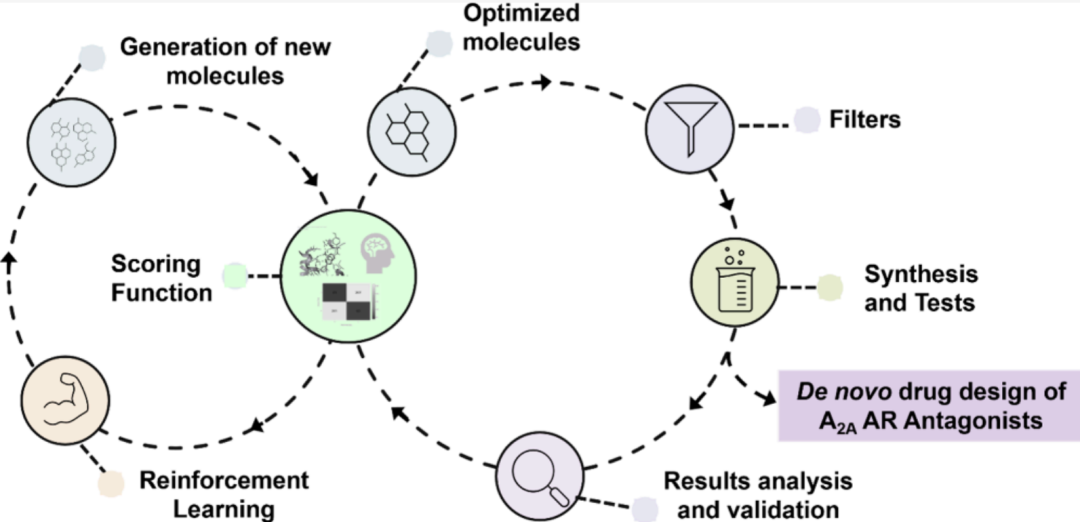
1 研究背景
1.1 早期药物发现中AI从头设计的定位
早期药物发现(EDD)阶段普遍面临高成本、高失败率、专利空间受限(经典筛选依赖已公开化合物库)等结构性问题。生成式AI(de novo design)被视为缓解这些问题的潜在路径,但其应用仍存在明显短板:对高质量、大规模靶点数据的依赖性强;现有方法普遍是靶点特异性而非通用性的;AI输出仍需研究者验证与解读,尚不能替代人类专业判断。
文献中绝大多数AI辅助EDD工作集中于基于配体(ligand-based)的超大库虚拟筛选,而真正在生成阶段就引入基于结构(structure-based)组件(即对接打分作为强化学习目标的一部分)的工作仍相对有限。本研究的核心定位,正是系统评估"在生成阶段嵌入结构信息"这一策略的实际效果与边界条件。
1.2 选择A2A腺苷受体作为模型靶点的依据
A2A AR属于G蛋白偶联受体(GPCR)腺苷受体家族成员(与A1、A2B、A3 AR同源),在血管扩张、肿瘤免疫抑制以及神经炎症/神经退行性疾病中均发挥关键作用。其拮抗剂伊曲茶碱(istradefylline)已获批用于帕金森病治疗,该受体在阿尔茨海默病、肌萎缩侧索硬化(ALS)、亨廷顿病中也被广泛认为具有治疗潜力。
选择该靶点的关键原因在于其"数据基础设施"的完备性:已发表配体数量大、活性/亲和力数据质量及一致性高,同时已公开了数量可观的共结晶结构(本研究药效团建模时纳入了58个拮抗剂结合状态的A2A AR X射线晶体结构)。这使A2A AR成为评估生成式AI方法学优劣的理想"压力测试"靶点——若方法在数据条件最优的情形下仍频繁失败,则在数据稀缺的"孤儿靶点"上的适用性将面临更严峻的挑战。
1.3 与既往工作的方法学差异
本研究在引言部分明确将自身与 Thomas 等人 2025 年发表于 Nat. Commun. 的同靶点研究(参考文献17)进行了区分:在 Thomas 等的工作中,基于结构对接是强化学习的主要优化目标,药物相似性质与特定残基相互作用处于次要地位;而本研究采取相反的实验设计——先以三组分等权重的"标准"配置进行第一轮生成与验证,在观察到失败后,有意识地将结构约束(特定残基相互作用)提升为主要驱动力,从而系统性地考察评分函数参数化对最终产出质量的因果性影响。
2 计算方法体系
2.1 数据整理与数据集构建
数据整理通过 KNIME 4.7.2 工作流完成,核心步骤包括:基于 ChemAxon 与 RDKit 的结构标准化(清洗结构、中和电荷、生成最可能互变异构体)→ 数据集精炼(按检测类型聚合、活性值转换为对数尺度、去重时保留最高活性记录)→ 输出 SDF/CSV 格式用于建模。
四个腺苷受体亚型的原始数据规模如下:
A2A AR模型额外纳入了一个74个化合物的外部验证集(数据采集截止日期之后新发表于ChEMBL的化合物),用于独立评估模型的前瞻预测能力。
2.2 配体基础组件:QSAR活性预测模型
第一轮建模采用 Evotec 自动化建模管线,系统比较了多种建模配置:
• 活性阈值(pKi/pIC50 = 6、7、8三档) • 二分类 vs. 三分类(后者引入"中等活性"不确定区间) • 检测类型组合(结合实验 vs. 功能实验;Ki vs. IC50)
分子描述符采用 MACCS指纹 联合 18个理化性质描述符(TPSA、ALogP、分子量、FSP3、重原子数、原子数、氢键供体数、芳香杂环数、杂原子数、可旋转键数、芳香环数、芳香碳环数、脂肪杂环数、脂肪环数、饱和杂环数、饱和环数、脂肪碳环数、饱和碳环数,均由 RDKit v2021.3.5 计算)。随机森林(RF)与支持向量机(SVM)模型均通过 scikit-learn 0.21.3 训练,五折交叉验证后保留性能最优者,超参数通过网格搜索优化。
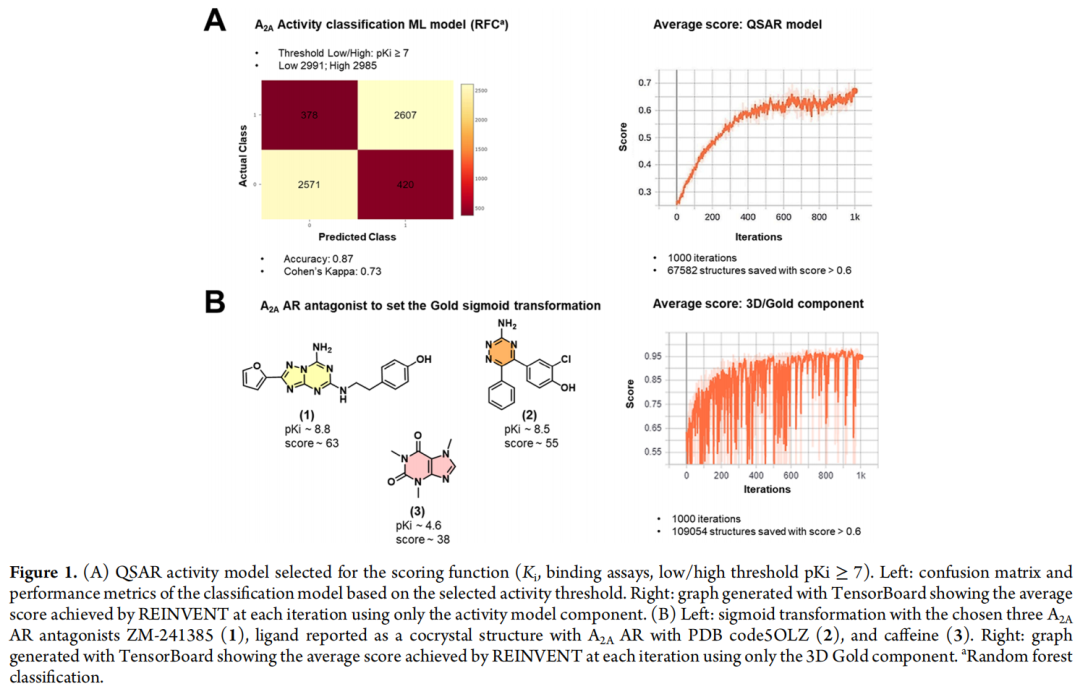
最终生产模型(用于评分函数的离散过滤器)为基于 pKi≥7 阈值的二分类模型:预测为"活性"赋分1,预测为"非活性"赋分0。性能指标:
外部验证后,该74个化合物被并入训练集以生成最终生产模型。
第二轮建模(用于评分函数重新校准):在第一轮9个化合物全部失活的反馈后,团队采用 Evotec 开发的另一套自动化流程(基于 RDKit 2021.03.1 + pycaret 2.3.10),描述符体系升级为 Morgan指纹(2048位)+ Atom Pair指纹(1024位)+ Topological RDKit指纹(1024位)+ MACCS(167位)+ RDKit分子描述符(208个理化/结构/拓扑性质) 的拼接特征;采用85:15训练-测试集划分(回归任务)或分层划分(分类任务),10折交叉验证优化超参数,最终在全部数据上重新训练。这一调整的核心意图是将"失活三唑"的新数据反馈回模型,引导后续生成偏离已证实的无效化学空间。
2.3 结构基础组件:分子对接打分
采用 GOLD程序(GoldScore打分函数),对接靶点为A2A AR的拮抗剂结合晶体结构(PDB: 4EIY)。受体结构经 MOE2022 套件预处理:去除融合蛋白apocytochrome b(562)RIL(替代了第三胞内环ICL3),并通过 Loop Modeler 基于知识库方法重建ICL3(Lys209–Gly218);补全缺失原子,N/C端以乙酰基/N-甲基酰胺基团封端;通过 Protonate3D 在生理pH(~7.4)下分配最可能质子化状态,Amber14:EHT力场最小化新增氢原子;保留晶体学钠离子(作为负性别构调节剂)及其溶剂化层的3个水分子。对接搜索空间限定在以Asn253与Phe168质心为中心的11Å球形区域内,每个化合物生成10个对接姿势。
为将原始GOLD打分映射至0–1区间,采用了基于三个参照拮抗剂的S型(sigmoid)转换校准:
据此设定:对接打分 <50 → 评分0;50–65 → 0至1连续映射;>65 → 评分1。该校准策略的合理性建立在"高亲和力配体应获得高对接打分"的假设之上,但也正因为这一映射区间较宽(50–65),为后续失败埋下了隐患——较宽的容许区间意味着大量"塞得进口袋但缺乏关键相互作用"的分子同样可以获得高分。
2.4 药代动力学组件:CNS-MPO评分
采用 Wager 等提出的 中枢神经系统多参数优化评分(CNS-MPO),综合评估6项理化性质以预测血脑屏障(BBB)穿透能力,原始量表为0–6分,>4分被视为最佳候选。评分函数中采用升序S型转换:CNS-MPO <3 → 0;3–4 → 0至1连续映射;>4 → 1。后续过滤阶段还引入了独立的 BBB Score(基于芳香环数、重原子数、MWHBN复合描述符、tPSA、生理pH下pKa等不同理化维度),作为CNS-MPO的交叉验证指标。
2.5 REINVENT生成框架与强化学习设置
REINVENT 是基于循环神经网络(RNN)+ 强化学习的生成式分子设计平台,核心特性是在生成过程中持续执行多参数优化(MPO)。本研究采用 Evotec 定制版(基于公开版本 REINVENT 3.2),关键设置包括:
• Inception策略:以三个文献已知拮抗剂(ZM-241385、PDB 5OLZ配体、咖啡因)作为初始种子,引导模型早期探索方向; • 多样性过滤器(Diversity Filter):基于相同Murcko骨架、最低相似度阈值0.4,防止生成过程收敛到单一狭窄区域; • 强化学习算法:第一轮采用标准策略,第二轮引入 Augmented Hill Climb(Evotec在原版REINVENT基础上新增的算法,用于加速多参数优化收敛)。
2.6 复合评分函数的整合逻辑
三个组分(配体活性、结构对接、CNS渗透性)各自归一化至0–1区间后取算术平均,总分>0.6的分子被视为满足设计标准并保留为模型输出。研究团队系统比较了四种评分函数配置(单独QSAR、单独3D/Gold、两者等权重组合、三者等权重组合)在1000轮迭代下的优化轨迹与产出规模,以验证生成模型对各单一组分及组合目标的优化能力。
3 第一轮生成:标准等权重评分函数
3.1 生成阶段产出统计
四种评分函数配置各运行1000轮迭代,以平均分>0.6为采集阈值:
| 四套运行合计 | 365,577 |
经KNIME后处理(去重、理化性质计算、Tanimoto相似度计算、指纹聚类)后,最终保留 365,321个独立分子,作为后续两套过滤策略共同的起点数据集。
3.2 多级后处理过滤管线(标准策略)
| 438 | ||
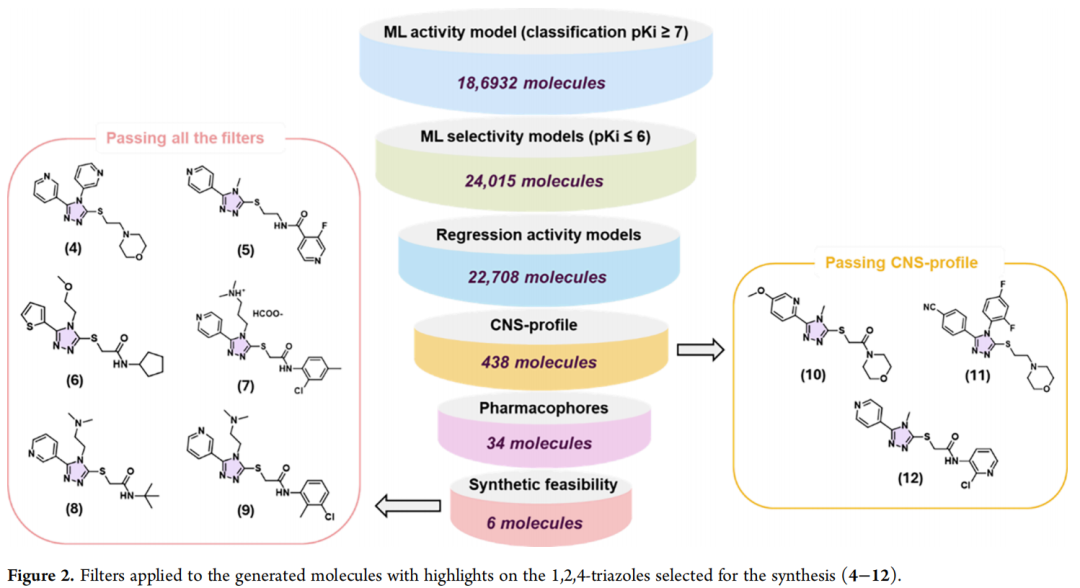
值得注意的是,第4步(CNS渗透性过滤)是整条管线中淘汰率最高的环节(从22,708骤降至438),说明在该评分体系下,"活性可预测"与"具备CNS成药性"之间存在显著的化学空间错配——这也从侧面解释了为何最终幸存的分子集中于结构高度同质化的骨架(1,2,4-三唑)。
在438个分子中,通过来源追溯发现 185个(约42%) 来自"QSAR + Gold + CNS-MPO"三组分组合运行,即三组分协同优化的策略在通过CNS筛选方面表现相对突出。
3.3 基于药效团与对接姿势的结构筛选
438个分子经 OpenEye 套件(Tautomers/Omega/Fixpka工具)处理互变异构体、3D构象与生理pH电离状态后,得到 505个结构,使用GOLD–GoldScore对接于A2A AR(PDB: 4EIY),共生成 5,050个对接姿势(每化合物10个)。
基于58个拮抗剂结合状态A2A AR晶体结构构建的共识药效团模型,定义了三级递进过滤标准:
| P1 | |||
| P2 | |||
| P3 | 34 |
最终34个分子经SciFinder新颖性与合成可行性检索,发现 23/34(约68%)为1,2,4-三唑骨架(3、4、5位取代)——这一高度集中的结构偏好直接反映出REINVENT在当前评分函数引导下的化学空间收敛特征。
3.4 化合物4–12的合成与药理学评价
初选6个三唑衍生物(化合物4–9)合成后,团队对该骨架在CNS-profile过滤阶段被淘汰的同类物进行回溯分析,基于合成可行性补选3个(化合物10–12),最终共9个化合物进入湿实验验证。化合物结构均为4,5-二芳基(或二吡啶基)-1,2,4-三唑-3-硫醇衍生物,3位通过硫醚键连接吗啡啉乙基、二甲氨基烷基、酰胺类等多样化侧链。
全部9个化合物经放射性配体竞争结合实验测定,A2A AR的Ki值均大于30,000 nM,即无活性。
研究团队特别指出,失活化合物并非集中来自单一评分函数配置(4个来自三组分组合,5个来自双组分组合),说明失活的根源不在于评分函数权重的具体取值,而在于评分体系本身未能编码某个对活性至关重要的结构特征。
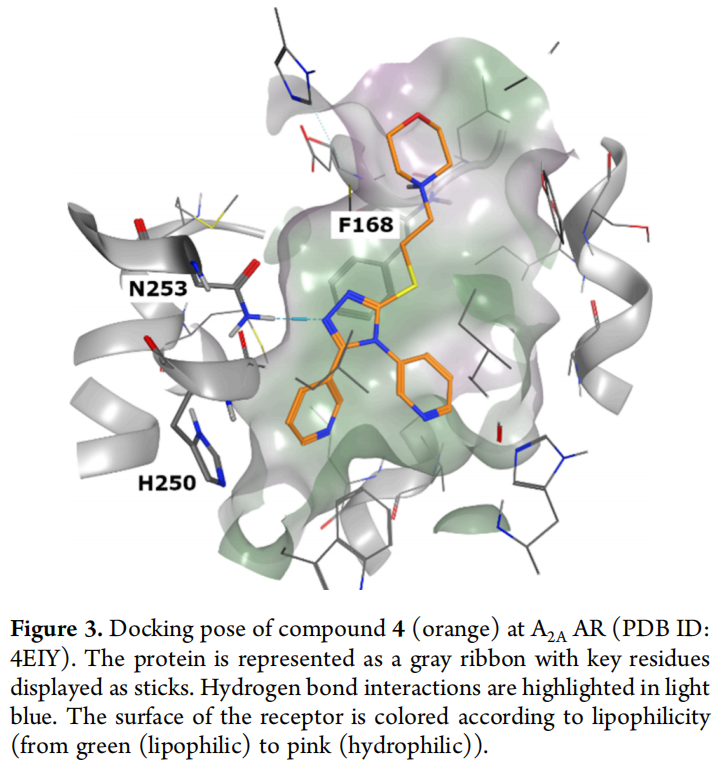
3.5 失活归因:结合模式复盘分析
以化合物4为代表的对接姿势复盘揭示了关键问题(见原文图3):三唑芳香环与Phe168形成π-π堆积;三唑2位氮原子与Asn253形成单一氢键(而非双齿氢键);5-芳基取代基深入口袋底部,与His250形成T形π-π相互作用;4-芳基取代基朝向TM1-2跨膜区;3位取代基则伸向受体细胞外开口区域。
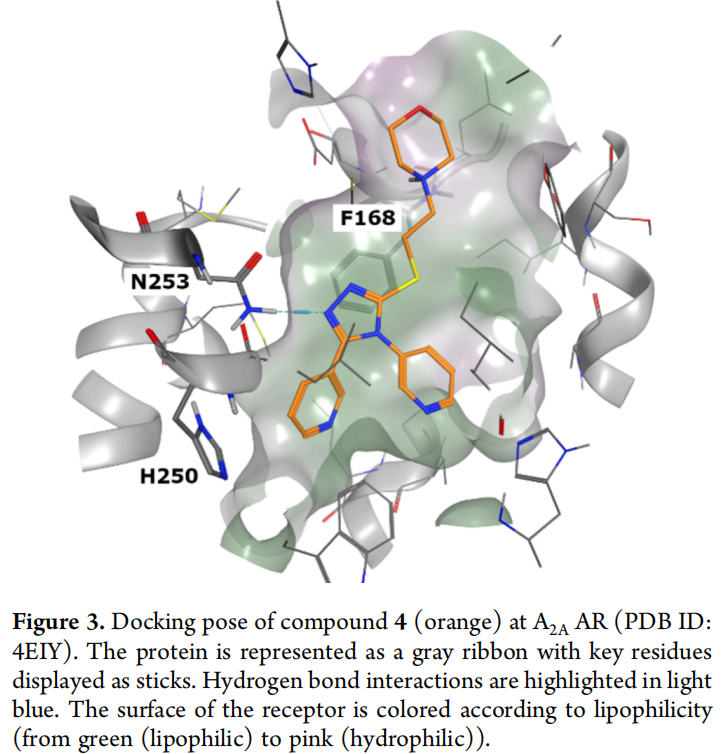
这一结合模式与文献报道的多数强效A2A拮抗剂存在本质差异:后者普遍依赖与Asn253形成的双齿氢键作为高亲和力的结构基础。研究结论明确指向:该相互作用未被纳入对接打分的隐式判据,也未在初次药效团设计中被设为强制性筛选标准(双齿氢键过滤虽被纳入P1后续步骤,但因合格分子过少且存在其他问题而被放弃执行),致使整条"事后过滤"管线无法弥补生成阶段就已缺失的关键化学特征。
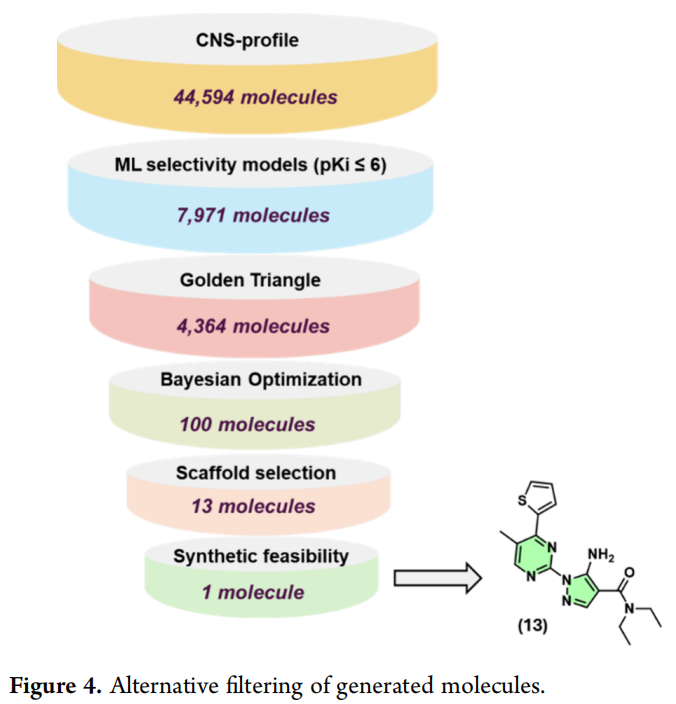
4 替代过滤策略:贝叶斯优化驱动的二次数据挖掘
4.1 过滤逻辑重构
为验证失败根源是否在于过滤策略而非生成阶段本身,团队对同一批365,321个分子(与3.1节起点完全相同)应用了顺序与类型均不同的过滤管线:
4.2 贝叶斯优化方法学
贝叶斯优化是一种基于"数据-标签对"训练代理模型(通常为高斯过程模型)的监督学习排序方法,代理模型输出预测均值与标准差,经采集函数转化为对未知点的排序依据。本研究中,Evotec开发的命令行工具采用以下设置:
• 模型:贝叶斯优化(BO) • 描述符:CDDD(Continuous and Data-Driven Descriptors,基于化学表示互译学习得到的连续型描述符) • 参考数据集:5,934个已知pKi值的A2A拮抗剂 • 采集函数:Expected Improvement(EI),jitter=0.01 • 优化目标:A2A拮抗剂pKi • 高斯过程引擎:botorch
该方法对4,364个分子进行排序,取前100个用于后续分析(Table S6)。
4.3 跨管线比较分析与候选确定
将BO排序前100个分子与标准管线产出的438个分子(同样排除三唑骨架)在 DataWarrior 中进行自动SAR分析,识别两个子集间共享的优势核心环系,最终确定13个高优先级分子进入合成评估(Figure S5)。
其中,吡唑-嘧啶骨架的化合物13因带有游离氨基(理论上具备形成Asn253双齿氢键的潜力)被特别关注——值得说明的是,该化合物并未通过标准的分子建模过滤管线入选,原因是其预测对接姿势实际上完全未显示与Asn253的氢键相互作用;研究者基于结构推理(而非对接打分本身)判断其具有潜在价值,决定将其纳入合成计划,这体现出人类专家判断在AI输出后处理阶段仍不可替代的现实。
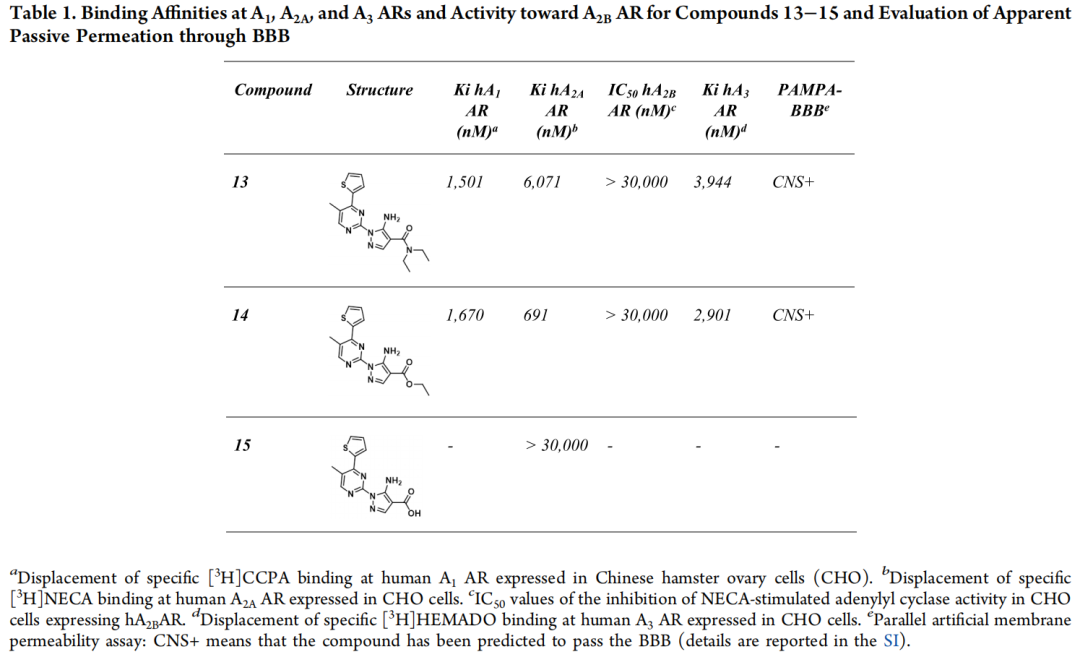
4.4 化合物13–15的合成与药理学评价
化合物13(5-氨基-N,N-二乙基-1-(5-甲基-4-(噻吩-2-基)嘧啶-2-基)-1H-吡唑-4-甲酰胺)及其两个合成中间体——乙酯化合物14、羧酸化合物15——的完整药理学数据如下:
| 13 | 6,071 | ||||
| 14 | 691 | ||||
| 15 |
关键发现:
1. 化合物13确证有效(Ki = 6.07 μM),是这一替代策略唯一、也是全文唯一来自全新化学骨架的实验验证命中分子; 2. 合成中间体14的活性反而优于终产物(Ki = 691 nM,纳摩尔级),提示侧链末端的二乙基酰胺基团相对乙酯基团在该位点可能存在不利的立体或电性效应,这是一个具有SAR教学价值的"细微结构改动导致活性悬崖"的实例; 3. 羧酸中间体15完全失活,进一步印证该侧链区域对静电/氢键性质高度敏感; 4. 化合物13与14均经PAMPA-BBB实验确证可穿透血脑屏障,验证了CNS-MPO组分在评分函数与过滤体系中的预测有效性。
5 第二轮生成:结构约束驱动的评分函数优化
5.1 评分函数修订的理论依据
基于3.5节的归因分析,研究假设"与Asn253形成双齿氢键"是A2A拮抗剂活性的必要结构特征(该假设有多篇独立文献支持)。因此第二轮的核心策略是将这一假设从"事后过滤标准"前移为"生成阶段的强化学习优化目标"。
5.2 关键参数调整
| 缺失则施加惩罚分 | ||
| 收紧至55–65 | ||
| 提升至5或10 | ||
| Augmented Hill Climb | ||
| 纳入第一轮失活三唑数据重新训练 |
研究同时讨论了一种正在兴起的替代范式——基于Pareto多目标优化的评分策略(参考文献82–84),相较于本研究采用的加权均值评分函数,Pareto方法理论上能更好地权衡多目标间的取舍。但本研究有意选择"largely as is"地使用REINVENT原生框架,目的是更纯粹地评估该工具本身在生成与后处理两个阶段的能力边界,而非引入额外的优化范式变量。
5.3 生成阶段产出统计
分别以权重5、权重10进行两组各1000轮独立迭代:
| 合计 | 117,409 |
经相同的KNIME精炼流程后,保留 117,386个分子(精炼损耗极小,与第一轮365,577→365,321的比例基本一致)。
5.4 第二轮过滤管线
第二轮采用了更精简、聚焦的过滤策略(活性—CNS—结构三级核心标准),不再单独设置亚型选择性过滤步骤:
| 4个分子(17–20) |
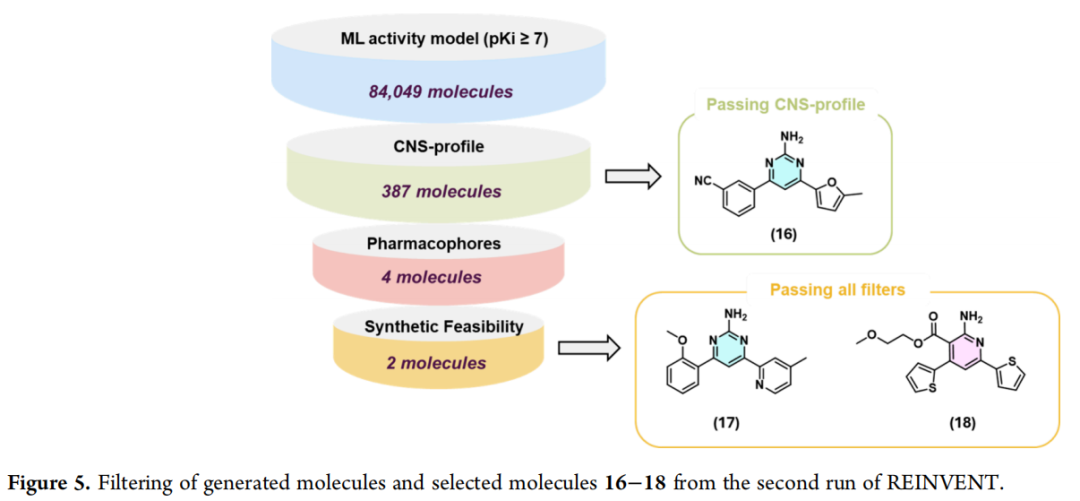
最终4个候选分子(17–20)的对接姿势均确认能与Asn253形成双齿氢键(由游离氨基与环内氮原子协同实现,涉及氨基嘧啶、氨基吡啶、氨基噻唑等骨架),中心芳香核与Phe168形成π-π堆积,芳基取代基靠近His250——即生成阶段的结构约束已在最终候选层面得到完整体现。
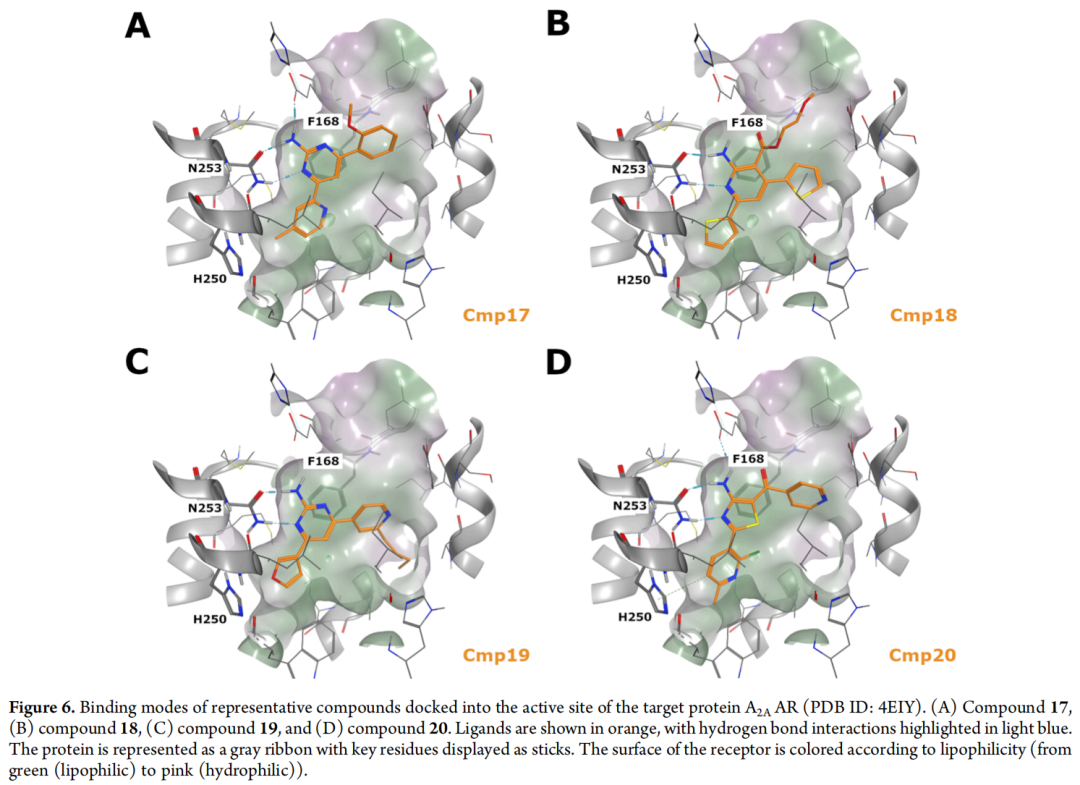
基于合成可行性考量,最终选择2-氨基嘧啶(17)与2-氨基吡啶(18)进行合成,并额外合成了与17结构相近、但未完全通过药效团过滤的类似物化合物16,作为构效关系(SAR)比较的参照分子。
5.5 化合物16–18的合成与药理学评价
| 16 | 179 | ||||
| 17 | 525 | ||||
| 18 | 521 |
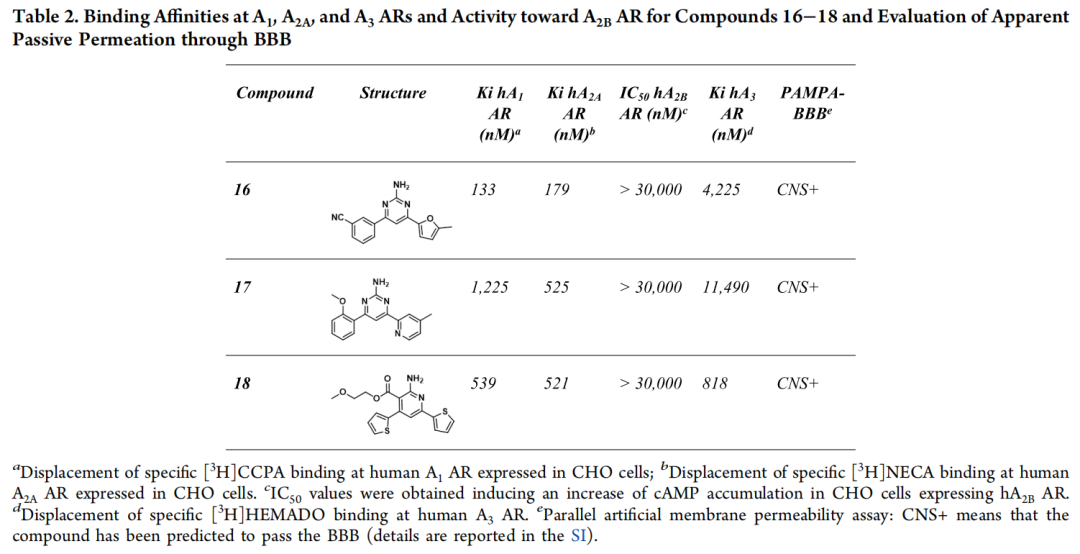
三个化合物全部确证有效,A2A AR亲和力落在179–525 nM区间(高纳摩尔至数百纳摩尔级),全部经PAMPA-BBB实验确证可穿透血脑屏障——实验成功率达到100%,且最佳活性(化合物16,Ki=179 nM)相较第一轮替代策略的最佳结果(6.07 μM)提升约34倍。
5.6 选择性谱型与化学新颖性分析
需要客观指出的局限性:三个化合物对hA1 AR与hA3 AR的选择性均不理想(尤其对hA1 AR),仅对hA2B AR表现出良好的选择性。研究坦诚说明,这一结果在预期之内,因为第二轮评分函数本身未设置亚型选择性过滤步骤——这是为聚焦验证"结构约束效果"而做出的实验设计简化,并非方法学缺陷,但也提示后续工作若以成药性为目标,仍需重新引入选择性优化组件。
化学新颖性方面,2-氨基嘧啶骨架的发现具有一定的标志性意义:对起始数据集的回溯分析显示,共106个分子含有2-氨基嘧啶核心,但绝大多数为四取代而非本研究的三取代模式;仅2个分子与生成结果高度相似(文献报道为A1拮抗剂)。值得关注的是,etrumadenant——一种目前处于II期临床(用于转移性结直肠癌)的双重A2A/A2B拮抗剂,其核心骨架同样为联芳基2-氨基嘧啶——并未出现在训练数据集中,但REINVENT在几乎没有直接先例的条件下生成了结构高度相关的化合物,这是本研究中AI生成能力的一个积极信号。
6 三轮实验综合数据汇总
为便于横向比较,下表汇总全部实验验证化合物的关键数据:
| 全新骨架 | ||||||
7 综合讨论
7.1 三轮实验定量对比
| 强制启用 | |||
7.2 Tanimoto相似度分析与"新颖性—命中率"权衡
通过Tanimoto相似度分析(原文Table S9)发现一个具有方法学意义的趋势:第一轮生成的化合物(包括失活三唑与有效化合物13)对所有参照数据库的相似度均低于0.61,即化学空间新颖性较高;而第二轮生成的化合物相似度评分明显更高,提示引入强结构约束后,生成模型更倾向于收敛到化学已知、结构成熟的母核区域。
这一发现揭示了AI从头设计中一个值得长期关注的内在张力:结构约束越精确(越接近已知有效相互作用模式),实验命中率越高,但生成结果的化学新颖性也越容易随之下降。在全部实验验证的命中化合物中,仅化合物13代表真正意义上的全新化学型(novel chemotype);第二轮的16–18虽然活性更优、实验成功率更高,但更接近已被探索过的化学motif。
7.3 评分函数设计的核心作用:不止是技术细节
本研究最具普适价值的方法学结论是:评分函数的参数化方式与生成后过滤管线的设计逻辑,对最终实验结果具有决定性影响,且二者的影响可以相互独立。证据链如下:
• 同一批365,321个生成分子,经标准过滤管线得到全部失活的9个化合物;经替代过滤管线(贝叶斯优化排序+跨数据集结构比较)却能挖掘出有效命中化合物13。这说明生成阶段本身已经产出了部分潜在有效分子,但标准的"基于对接姿势的药效团过滤"未能识别出它们——换言之,过滤管线的设计缺陷,而非生成模型能力不足,是Run1标准策略失败的直接原因之一。 • 当把"双齿氢键"这一已知关键判据直接前移至生成阶段的强化学习目标(而非仅作为下游过滤条件)后,Run2的实验成功率从0%跃升至100%,印证了**"硬知识应尽早注入生成循环,而非留待后处理"**这一设计原则的有效性。
7.4 局限性:数据丰富靶点与"孤儿靶点"的适用性差异
研究明确指出,本方法体系高度依赖A2A AR本身的"数据丰厚性"——大量已知配体、丰富的共结晶结构、明确的关键残基相互作用文献支持,这些都是构建有效QSAR模型、校准对接打分、定义可靠药效团约束的前提条件。对于结构信息匮乏、配体数据稀少的"孤儿靶点",当前方法体系中的多数组件(尤其是QSAR分类模型与基于已知关键相互作用的药效团约束)将难以构建或缺乏可靠性——而这类靶点恰恰是计算驱动从头设计最需要发挥价值、也最难以落地的应用场景,构成了该方法学路径当前最主要的现实瓶颈。
7.5 对方法学实践的启示
综合全文,可以提炼出若干对从事AI辅助药物设计的研究者具有直接参考价值的实践建议:
1. 评分函数中各组分的权重不应默认等权处理,而应基于对靶点结构生物学认知(如已知的关键残基相互作用)进行有依据的差异化设置; 2. 结构约束(如特定残基的氢键模式)应尽可能编码为强化学习阶段的显式优化目标或惩罚项,而非仅作为生成后的过滤标准——后者只能从已生成的有限候选中"挑剩下的",无法引导生成模型主动探索符合约束的化学空间; 3. 生成结果的后处理过滤策略本身应被视为一个独立的、可系统优化的实验变量——同一份生成产出,不同的过滤逻辑可能导致截然不同的结论,因此在评估生成模型"是否失败"之前,应首先排除过滤管线设计本身的问题; 4. 机器学习排序方法(如贝叶斯优化)可作为传统基于规则/药效团过滤的有效补充,尤其适合在传统方法因结构特征定义过严而漏判潜在候选时发挥作用; 5. 应为"新颖性—命中率"权衡设定明确的项目优先级:若以快速验证可成药性为目标,适度收紧结构约束是合理选择;若以专利布局或全新化学型探索为目标,则需要在评分函数设计中保留更大的化学空间自由度,并接受相应的实验失败率。
总结
本研究通过两轮REINVENT生成实验与两套差异化的过滤策略,系统性地证明了AI从头药物设计中评分函数参数化与后处理过滤管线设计对最终实验结果具有决定性的、且相互独立的影响。标准等权重评分函数搭配基于药效团的过滤策略导致9个合成化合物全部失活;通过贝叶斯优化重构过滤逻辑后,从同一批生成分子中挖掘出1个微摩尔级全新骨架命中化合物(13);将失败归因(Asn253双齿氢键缺失)直接编码为第二轮评分函数的结构约束后,3个合成化合物实现100%的实验命中率,活性提升至高纳摩尔级,且均预测可穿透血脑屏障。
这一系列结果共同表明,成功的生成式从头设计高度依赖于将靶点特异性的结构知识系统性地整合进强化学习的优化目标,而非仅仅依赖更大规模的生成产出或更复杂的下游过滤。同时,该方法体系对数据丰富靶点的适用性已得到较充分验证,但对结构与配体数据匮乏的靶点的可推广性,仍是该领域亟待解决的核心挑战之一。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢