
目录
0 引言
1 金属配位几何与蛋白骨架拓扑的设计
2 金属离子选择性的结合设计
3 功能性金属结合位点的设计
4 参考文献
0 引言
金属结合蛋白能够参与多种生命过程,而金属在其中扮演着至关重要的角色。早在1996年,Holm等人就总结了金属结合位点的五大功能1。分别是:稳定蛋白质结构,调节蛋白质的构象;充当催化活性中心,辅助活化反应底物;传递电子;结合或运输小分子,比如参与氧气运输;储存金属离子,包括金属离子的摄取、缓冲和释放。不同类型的金属结合位点具有特异的结构特征,并赋予蛋白质多样的生理功能。而实现具有特定功能的金属结合蛋白设计,也成为蛋白质设计领域一个富有挑战的任务。
其中挑战性存在于如下几个方面。其一,如何协调金属结合位点所需的局部侧链配位几何与蛋白质二级结构及三级结构(即骨架拓扑几何)之间的关系2,以赋予蛋白质金属离子结合能力。这一问题与设计能够结合其他类型配体的蛋白质所面临的挑战具有相似性。不同的是,对于金属离子体系,还存在一种利用配位几何对称性的设计策略,即以满足特定几何约束的对称蛋白骨架作为设计起点。其二,如何通过对配位层残基(以及距离配位中心更远的其它残基)的精细调整,实现蛋白质对不同类型金属离子的区分。一个典型的例子(尽管并非蛋白质设计体系)就是钾离子通道对于钾离子和钠离子的选择性。离子半径更小的钠离子由于无法与通道侧链羰基氧紧密结合,其脱水能无法得到补偿,因此无法稳定占据并通过钾离子通道。其三,如何打破金属离子配位几何的对称性,引入除稳定蛋白质结构以外的功能特性。比如在金属蛋白酶的设计过程中,需要考虑蛋白质-金属-底物三元体系的互作,以及反应路径中体系构象的动态变化。从设计目标上来看,最后一个挑战一定程度上整合了前两个挑战,因此难度最大,同时也最富有吸引力。
本文,我们将基于蛋白质设计方法的基本原理、以上述三个挑战为主线,对一些典型的金属结合蛋白质设计案例进行简要的分类和介绍。
1 金属配位几何与蛋白骨架拓扑的设计
金属结合蛋白的设计包括金属配位几何的设计与蛋白骨架拓扑的设计。前者通常可以独立于具体蛋白骨架,通过结构数据库或量子化学计算确定理想配位几何;后者则意味着蛋白质骨架中存在若干位置,这些位置引入相应配位残基后,其侧链能够满足预先定义的金属配位几何约束。在早期大多数的金属结合蛋白设计过程中,通常采用的思路是:先固定理想配位几何,随后在天然蛋白骨架中进行匹配,最后再优化局部环境。随着深度学习的发展,借助生成式模型的设计思路也不断涌现。这些方案以定义好的金属结合基序作为约束直接生成蛋白骨架。
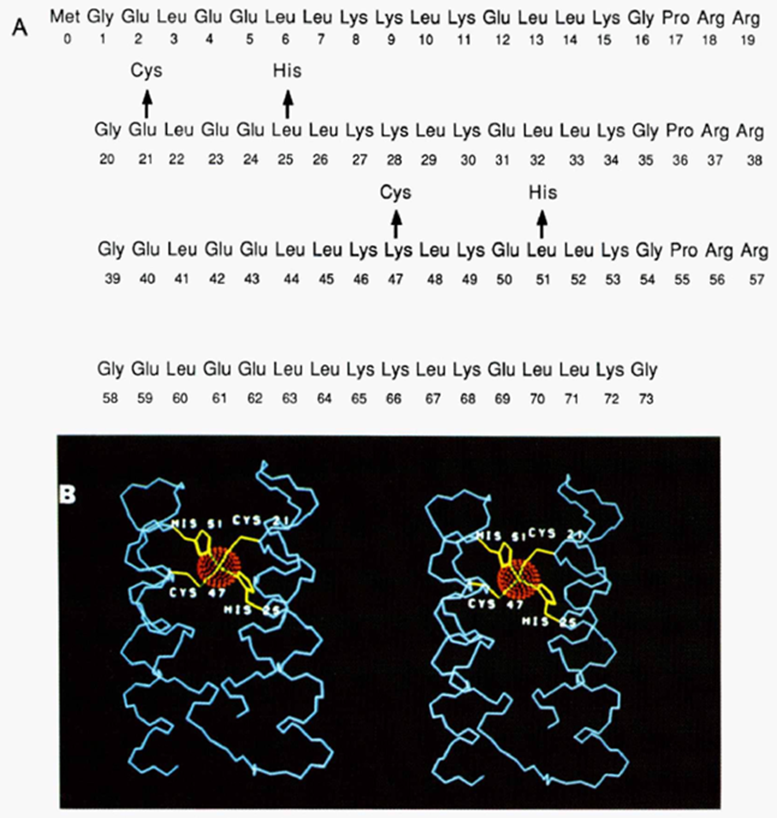
早期工作旨在构建一些三螺旋和四螺旋骨架,从而满足金属结合位点设计中的对称性要求。DeGrado课题组于1988年从头设计得到由α螺旋构成的单链四螺旋束蛋白(α4蛋白)3。通过在序列中周期性地引入疏水残基和亲水残基,蛋白能够自发地通过螺旋一侧的疏水作用聚集、通过螺旋另一侧的亲水作用稳定其在水溶液中的结构。在后续的延伸工作中(1990年)4,作者进一步利用α4蛋白骨架,搜索螺旋束界面中哪些位置能够支持两个Cys和两个His,并保证侧链硫原子和氮原子能够位于四面体的顶点处。最终搜索得到一个满足条件的位点,并验证了引入Cys和His突变的蛋白具有锌离子结合能力。这是最早报道的从头设计的金属结合蛋白。
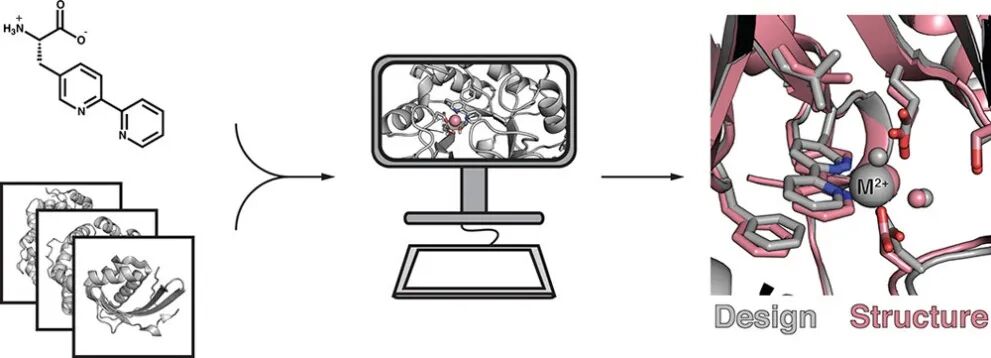
Baker课题组还通过引入非天然氨基酸拓展了配位残基的设计空间5。作者声称引入(2,2′-联吡啶-5-基)丙氨酸(Bpy-Ala)在设计层面具有两方面的优势。其一,Bpy-Ala所含有的两个吡啶氮原子具有预组织的空间排布,可直接形成双齿配体。这相比于同时控制两个组氨酸侧链的咪唑氮来构建金属结合位点而言,设计难度大大降低。其二,Bpy-Ala本身不带电,因此可以用于设计位于蛋白质内部深埋区域的金属结合位点。Bpy-Ala被用于构建理想的金属配位几何(Theozyme),随后使用RosettaMatch算法扫描天然蛋白质的骨架结构,以获取满足几何条件的主链。
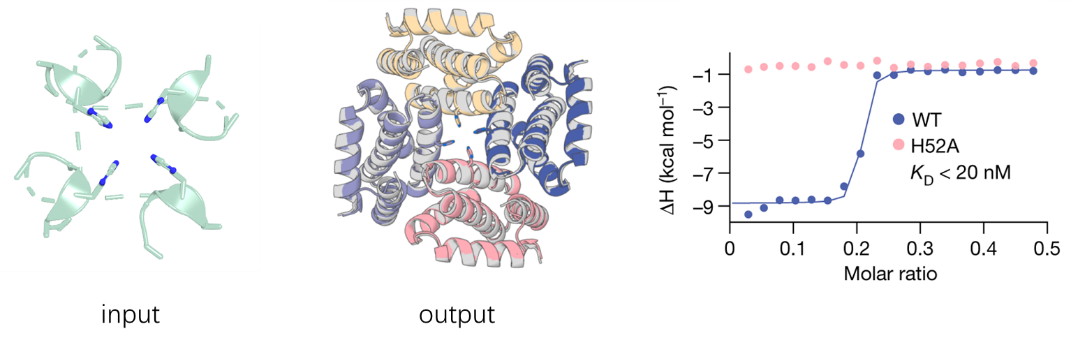
近年来,深度学习技术的发展为金属结合蛋白的设计开辟了新的道路。比如Baker课题组发展的RFdiffusion6,直接将金属配位几何作为约束条件嵌入蛋白骨架的生成过程。在这一框架下,作者描述了结合镍离子的对称四聚体的设计过程。用户首先需要提供已经按照目标对称性处理好的金属结合基序,同时指定目标蛋白满足C4对称性;随后使用ProteinMPNN设计序列、并使用AF2筛选设计结果;最终得到具有纳摩尔级亲和力的四聚体蛋白。相比于对称体系,针对非对称金属结合蛋白的生成式设计目前报道仍相对较少。
由此可见,金属配位几何的构建方法已较为成熟,而如何设计或筛选能够稳定承载预定义配位几何的蛋白骨架,则成为金属结合蛋白设计中的关键问题。目前多数工作都聚焦于具有稳定二级结构(尤其是α螺旋结构)的骨架设计中。上文提及的Bpy-Ala案例中,作者就在第二轮设计中加入了“Bpy-Ala必须位于稳定二级结构”这一约束,以避免第一轮设计中出现的“Bpy-Ala位于loop区、翻转进入溶液中”的现象。如何将金属结合蛋白的设计拓展至β折叠、混合折叠乃至天然无序区域,是值得进一步研究的问题。此外,对于apo态与holo态之间伴随显著构象重排或配位几何变化的金属结合蛋白,目前的设计策略仍然面临较大挑战。
2 金属离子选择性的结合设计
天然蛋白质对不同金属离子的亲和力主要由金属离子的电子性质决定。从配位化学角度来看,天然蛋白中的金属结合体系可大致分为三类:以镁离子、钙离子为代表的碱土金属,以锰离子、铁离子、钴离子、镍离子、铜离子和锌离子为代表的过渡金属,以及铁硫簇等金属辅基。这三类体系具有较为明确的配位偏好。根据硬软酸碱(HSAB)理论,碱土金属等偏“硬”的路易斯酸倾向于与Asp、Glu侧链羧基氧以及主链羰基氧等偏“硬”的配体结合;过渡金属离子的酸碱软硬性质分布较宽,从较硬的Mn(II)到较软的Cu(II)/Zn(II),整体呈现连续变化趋势,更倾向于与Cys侧链硫原子、His侧链咪唑氮等配体形成配位键;而铁硫簇由于具有预组装的无机硫骨架,其结合方式与单核金属离子存在明显差异,通常由多个Cys残基进行锚定。然而,在同一类金属离子内部,不同金属之间的配位偏好往往高度重叠。例如,一些二价过渡金属均能够与His、Cys及Asp/Glu等残基形成稳定配位,并具有相近的配位数和配位几何;而在相同的配位环境下,过渡金属遵循Irving-Williams规则,结合能力由小到大依次为Mn(II)、Fe(II)、Co(II)、Ni(II)、Cu(II)和Zn(II)。因此,实现金属离子选择性的计算设计变得尤为困难。
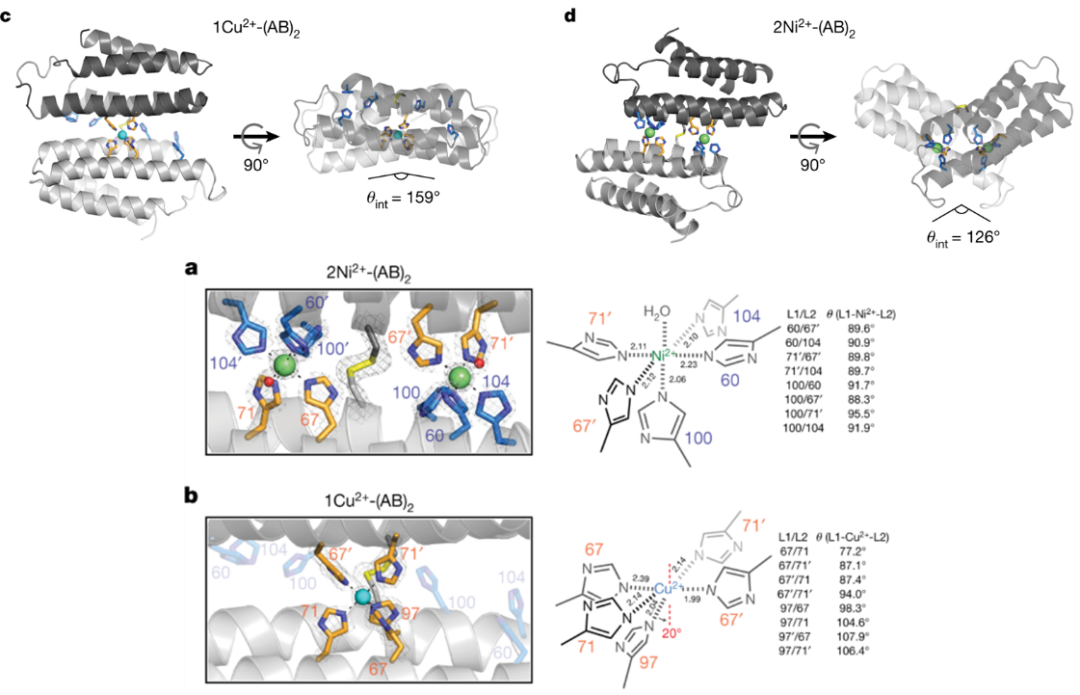
为了打破这一局限,Tezcan课题组利用MASCoT策略发展了一种可以克服Irving-Williams限制、实现对Ni(II)和Co(II)的选择性结合的方法7。MASCoT(metal active sites by covalent tethering)是Tezcan课题组此前发展的一种金属结合蛋白设计策略,即通过二硫键将两条蛋白链连接,蛋白质链能够围绕二硫键旋转,从而在互作界面形成多种不同的构象;这些构象为界面中的金属结合基序提供了多样的(而非预组织的)几何形状8。通过实验尝试,作者发现MASCoT在某一构象下(下图d)能够优先结合Co(II)、Ni(II),且这种选择性源自侧链形成的八面体配位几何的微妙差异。但总的来说,这一工作更像是“发现”了一个多构象蛋白结合不同金属离子的能力,而非是针对不同离子进行选择性区分的“有意设计”。
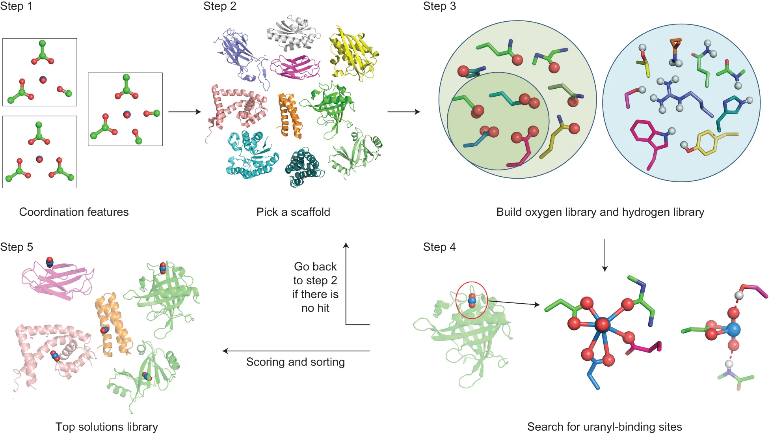
除了一般的过渡金属体系外,也有针对高价态锕系金属的选择性结合研究报道。在生物工程领域,何川课题组与来鲁华课题组合作报道过选择性结合铀酰的蛋白质设计工作9。铀在铀酰离子中通常以+6价形式存在,两端与氧原子形成线性配位,构成整体带+2电荷的线性三原子结构。除轴向的两个氧配体外,铀酰离子在其“赤道平面”上还能够进一步配位5或6个配体,从而形成五角或六角双锥配位构型。这种独特的配位特征使铀酰离子区别于大多数碱金属、碱土金属及过渡金属离子,表现出显著不同的结构化学性质。尽管此前存在一些铀酰结合基序的设计工作,但迄今尚无体系能够实现对铀酰相对于碳酸盐及其他金属离子的高选择性结合。作者通过在 PDB 数据库中筛选能够容纳上述配位几何的蛋白口袋(同时也将水分子纳入考虑),并结合定点突变优化,最终设计得到蛋白 SUP。在 pH 8.9 条件下,SUP 对铀酰表现出飞摩尔(femtomolar)量级的结合亲和力(Kd)。在选择性方面,该蛋白对钙离子的选择性超过 10⁶,对铜离子的选择性约为 10³。相比之下,SUP 对结构相似的钒酰离子选择性相对较弱,提示其高特异性可能主要来源于铀酰独特的配位几何与电子结构特征。
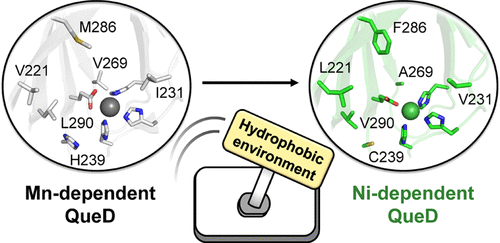
此外,还可以通过改造非配位层的残基调节蛋白质对金属类型的功能依赖性10。在Eom等人的研究中,作者以非血红素二氧化酶(quercetin dioxygenase,QueD)为模型体系,系统分析了不同金属依赖型变体,包括Mn依赖型与Ni依赖型QueD。尽管这些蛋白在整体折叠构象及第一配位层残基上高度保守并几乎完全重合,但其催化活性却呈现显著的金属特异性差异,表明金属选择性主要由第二配位层及其周围微环境所决定。通过序列分析,作者锁定了位于活性位点附近的关键非配位残基位点(如239位与290位),并通过多轮饱和突变与定向进化逐步实现了Mn依赖型QueD向Ni依赖型酶的转变。DFT计算进一步表明,第二配位层疏水环境的重构显著改变了底物芳香环在活性位点中的构象分布,使其更接近Ni中心所偏好的反应几何构型。尽管该研究并非直接针对蛋白质的金属结合能力进行改造,但它启示我们第二配位层在调控局部微环境的作用,并可能成为金属依赖性功能的关键因素。
以上设计工作表明,通过热力学控制的方法设计蛋白质对金属离子的选择性是可能的,但是这依赖于严苛的配位几何精度,而这种精度在复杂蛋白折叠体系中往往难以通过理性设计稳定复现。
3 功能性金属结合位点的设计
除了赋予蛋白质结合特定金属离子的能力外,功能设计还要求其能够借助金属离子实现催化反应、电子传递以及离子运输等关键生物功能。这类设计目标通常涉及多种不同的复合物构象状态,使得蛋白质设计从静态的几何匹配问题转变为动态的构象空间搜索问题。
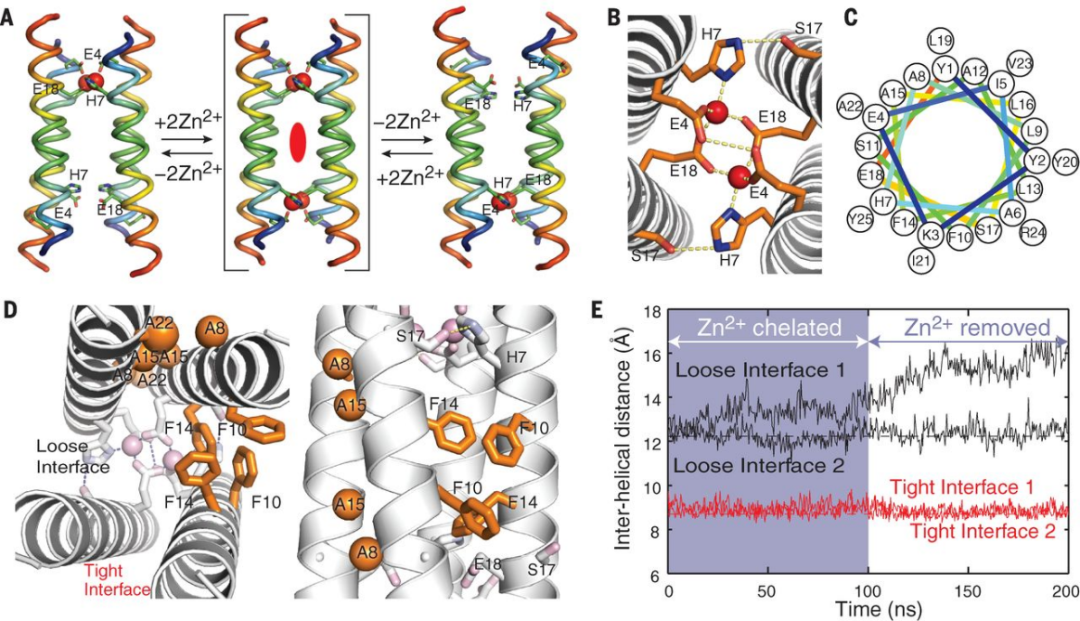
早在2014年,DeGrado课题组就报道了转运锌离子的膜蛋白设计11。天然转运蛋白被认为通过在两个或多个构象状态之间往复切换来实现其功能,这种机制被称为“交替通路机制”(alternating access mechanism)。考虑到目前的四螺旋束结构已被证明额能够运输质子、结合金属离子,于是作者从这一结构出发,尝试设计一个转运锌离子的跨膜蛋白。“交替通路机制”涉及向外开放(outward-facing)和向内开放(inward-facing)两个状态,前状态下结合位点暴露在膜外侧以结合底物,后状态下结合位点则暴露在内侧以释放底物。为实现此两种状态,作者设计了由多个苯丙氨酸构成的Loose interface作为摇摆轴(相对地,由多个丙氨酸构成的tight interface则用于固定构象)。锌离子的结合将拉近Loose interface的距离,从而形成一端关闭、一端开放的不对称状态。作者后续亦通过实验证明设计的蛋白能够选择性转运锌离子、钴离子(而不能转运钙离子),尽管其转运活性未达到天然转运蛋白的活性。
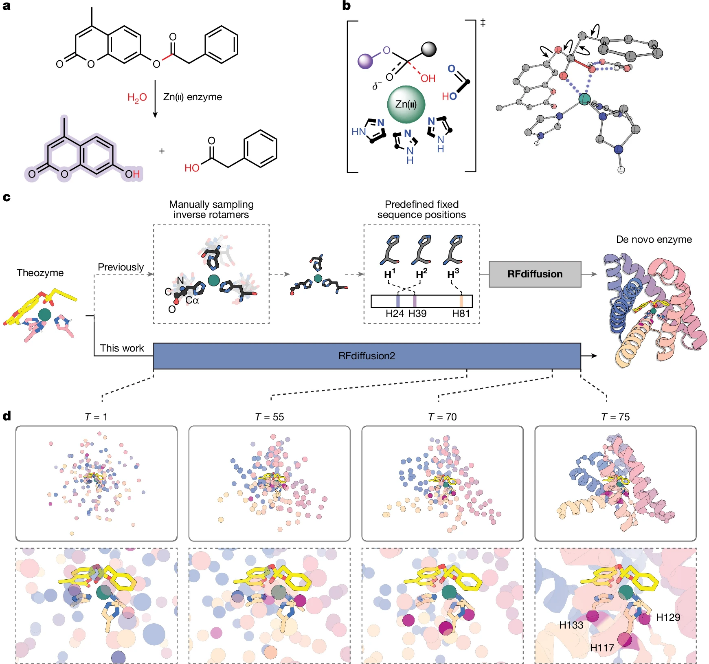
更具挑战性的金属酶设计任务在最近也实现了突破性进展。借助深度学习方法,Baker课题组报道了针对一种荧光酯(4MU-PA)水解反应的金属水解酶的计算设计12。作者首先利用DFT计算了Zn(II)-OH亲核进攻底物酯键的过渡态几何构型,得到一个理论酶(Theozyme),包含“最小活性中心“的3D坐标(一个锌离子、3个组氨酸的咪唑环、1个底物分子)。随后直接将理论酶输入至RFdiffusion2模型13中,得到设计好的酶骨架结构。此前的RFdiffusion对输入条件存在一些限制,比如只能以主链结构表示理论酶中的残基、需要给出残基在主链上的位置等等,这些限制既增加了计算耗时也损失了理论酶中的原子级motif信息。RFdiffusion2则通过移除部分残基在序列中的索引等训练方式克服了上述局限。其次,作者使用ProteinMPNN和AlphaFold2进行序列设计和筛选,并使用PLACER模型14评估活性中心是否高度预组织。作者从96组设计中筛选得到ZETA_1蛋白,该蛋白具有远超以往设计金属水解酶的活性(kcat/KM为16000)。通过突变实验,作者进一步修正了此前的理论酶结构,在第二轮筛选中得到ZETA_2,其活性得到进一步提升(kcat/KM为53000)。
金属离子以其独特的电子结构、配位化学和物理性质,大大拓展了化学反应的边界。早期Arnold课题组就通过对P450的定向进化和工程化改造,成功将这一天然血红素酶的催化空间拓展至非天然反应,实现了诸如卡宾/氮烯转移、碳-氢键精准活化以及非天然碳-硅/碳-硼键形成等极具挑战的化学转化。如今,随着深度学习技术的发展,从头设计功能性金属酶正在摆脱对天然同源骨架的依赖,实现了基于量子化学过渡态几何约束的原子级精准生成与功能定制。我们期待,在不久的将来,人工金属结合蛋白能够彻底打破自然演化的束缚,在绿色智能制造、极端环境生物修复以及新型功能材料开发等领域释放出颠覆性的应用价值。

4 参考文献
1.Holm, R. H., Kennepohl, P. & Solomon, E. I. Structural and Functional Aspects of Metal Sites in Biology. Chemical Reviews 96, 2239–2314 (1996).
2.Chalkley, M. J., Mann, S. I. & DeGrado, W. F. De novo metalloprotein design. Nature Reviews Chemistry 6, 31–50 (2022).
3.Regan, L. & DeGrado, W. F. Characterization of a Helical Protein Designed from First Principles. Science 241, 976–978 (1988).
4.Regan, L. & Clarke, N. D. A tetrahedral zinc(II)-binding site introduced into a designed protein. Biochemistry 29, 10878–10883 (1990).
5.Mills, J. H. et al. Computational Design of an Unnatural Amino Acid Dependent Metalloprotein with Atomic Level Accuracy. Journal of the American Chemical Society 135, 13393–13399 (2013).
6.Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
7.Choi, T. S. & Tezcan, F. A. Overcoming universal restrictions on metal selectivity by protein design. Nature 603, 522–527 (2022).
8.Rittle, J., Field, M. J., Green, M. T. & Tezcan, F. A. An efficient, step-economical strategy for the design of functional metalloproteins. Nature Chemistry 11, 434–441 (2019).
9.Zhou, L. et al. A protein engineered to bind uranyl selectively and with femtomolar affinity. Nature Chemistry 6, 236–241 (2014).
10.Eom, H., Cao, Y., Kim, H., de Visser, S. P. & Song, W. J. Underlying Role of Hydrophobic Environments in Tuning Metal Elements for Efficient Enzyme Catalysis. Journal of the American Chemical Society 145, 5880–5887 (2023).
11.Joh, N. H. et al. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 346, 1520–1524 (2014).
12.Kim, D. et al. Computational design of metallohydrolases. Nature 649, 246–253 (2026).
13.Ahern, W. et al. Atom-level enzyme active site scaffolding using RFdiffusion2. Nature Methods 23, 96–105 (2026).
14.Anishchenko, I. et al. Modeling protein–small molecule conformational ensembles with PLACER. Proceedings of the National Academy of Sciences 122, e2427161122 (2025).
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢