

2020 年 11 月举办的 CASP14 蛋白质结构预测国际竞赛上,AlphaFold 2 一战成名。凭借接近实验观测精度的蛋白质三维结构预测,AlphaFold 2 不仅一举攻克困扰生物学界长达半世纪之久的蛋白质折叠难题,还以惊艳的成果将其两位缔造者成功送到了四年后的诺贝尔化学奖舞台上。然而,AlphaFold 2 的成功也仅仅是一个开始,它如同一颗核弹般引爆了整个结构生物学领域,掀起了利用人工智能解析生物大分子空间构象的研究浪潮。
受 AlphaFold 2 在蛋白质领域巨大突破的启发,利用人工智能破解核糖核酸(RNA)的三维结构预测难题同样被寄予厚望。但目前主流算法落地时仍面临多重难以回避的现实瓶颈:其一,基于 Transformer 的 RNA 预测模型大多高度依赖多序列比对(multiple sequence alignments,MSA)提供的显性进化序列信息或间接利用生物语言模型得到的同源序列信息。而 RNA 碱基配对作用的等构特性,导致很难生成高质量、可靠的多序列比对结果;其二,多数主流方法未能充分挖掘 RNA 二级结构中碱基配对的关键信息,而碱基配对是决定 RNA 三维折叠形态的核心;其三,RNA 分子具备天然的构象柔性,同时存在多种稳定空间构象,现有算法大多仅能输出单一静态结果,无法还原 RNA 真实的动态构象集合。
针对上述挑战,美国弗吉尼亚理工大学的 Debswapna Bhattacharya 教授和其学生 Sumit Tarafder,合作开发出一款基于序列和碱基对条件的 SE(3) 等变流匹配(SE(3)-equivariant flow-matching model)模型 RNAbpFlow。该模型仅依靠 RNA 核苷酸序列与碱基配对信息就能生成完整全原子 RNA 构象集合,全程无需多序列比对、同源模板等进化信息,弥补了现有基于人工智能的 RNA 结构预测方法的多重短板。多项基准实验证实,引入碱基配对作为约束条件可大幅提升模型预测精度,在 RNA 拓扑结构采样和三维构象建模两项核心任务上,RNAbpFlow 整体表现均优于现有主流方法。
值得一提的是,在 RNAbpFlow 与 AlphaFold 3 的对照测试中,一组亮眼的数据揭开了最终博弈结果:在 CASP16 盲测实验中,14 条待测 RNA 靶标(≤200 nt)里,RNAbpFlow 能精准还原绝大多数靶标的全局折叠拓扑,共 12 条达到合格预测标准;相比之下,同等实验条件下 AlphaFold 3 仅 8 条靶标预测结构与天然构象匹配度达标。
相关研究成果以「RNAbpFlow: base pair-augmented SE(3) flow matching for conditional RNA 3D structure generation」为题,已发表于 Nature Methods。
研究亮点:
- 提出基于序列和碱基对条件的 SE(3) 等变流匹配模型 RNAbpFlow,弥补了现有 RNA 三维结构预测的多项短板
- 提出三通道碱基配对条件输入、核苷碱基中心表征、碱基配对专属辅助损失(bp2D 和 bp3D)三大创新,大幅提升碱基配对与三维结构的保真性
- 提出独立无重复的训练与测试集策略,采用多套权威基准与 CASP 盲测验证,综合性能领先现有主流算法

论文地址:
https://www.nature.com/articles/s41592-026-03128-4
独立去重训练集和测试集确保评估真实公平
为搭建自研模型 RNAbpFlow 并保证其评测结果真实可靠,本研究采用互相独立且无内容重复的训练集和测试集,并且针对不同测试基准配套专属模型权重,而非全程共用单一默认模型,以避免训练数据混入到评估环节造成结果失真。
模型开发及内部验证方面,研究采用 RNA3DB 数据集。这是一套十分适用于深度学习模型训练与内部基准测试的数据集,在序列层面与结构层面均具备非冗余特性。在本研究中,所用数据集选自 2024 年 4 月 26 日从蛋白质数据库(PDB)中解析得到的 RNA3DB 数据集版本,并沿用了原文给定的训练-测试划分方案,以筛选具有代表性的 RNA 序列用于实验。
为保证数据集仅包含高质量天然 RNA 结构,确保碱基配对信息准确,实验经过多层质控进行过滤,包括剔除单个核苷酸仅存单个原子的结构和混杂蛋白质残基的结构;截取连续实验序列,修正 FASTA 序列与三维结构不匹配问题,保护碱基配对完整性;删除天然结构中无碱基配对的序列等。在借助 RNAView 提取碱基配对信息,筛除长度 ≧ 20 个核苷酸的连续无配对片段的 RNA 链,并限定序列长度 30-200 后,最终得到含 560 条 RNA 序列的纯净训练集(剔除无法匹配 Rfam 家族的序列,进一步降低数据泄露风险)和含 48 条序列的纯净测试集。
值得一提的是,该部分数据划分完全遵循 RNA3DB 的非冗余拆分,训练和测试集不存在任何序列、结构重叠,以从根源上避免数据泄露。
在 RNAbpFlow 与 RNAJP 的对比实验中,研究采用了 RNAJP 研究中使用的含三向连接区的 22 条 RNA 数据集,并进行了筛选处理。研究人员先从该数据集中剔除多聚体结构,并将剩余序列与前述纯净训练集进行快速对比,去除冗余消除序列重复。最终处理后的数据集被缩减至 12 条。
在适配 CASP15 和 CASP16 国际竞赛方面,为了严格遵守竞赛盲测规则,同时保证与其他算法之间的公平对比,研究并未直接采用 RNA3DB 所提供的固有训练和测试集划分,而是从 RNA3DB 中重新整理并策划两套完全不相交、无重叠的新训练集。
对于 CASP15,由于 CASP15 竞赛首批待测 RNA 是 2022 年 5 月才对外公布的,研究人员在策划新训练集时只收录了 PDB 数据库中 2022 年 4 月之前上传的 RNA 序列(全部来自 RNA3DB),以保证模型训练时完全避开 CASP15 的待测 RNA,严格符合盲测规则。最终,训练集得到 731 条 RNA 序列,长度 30-784 个核苷酸。用于模型评估的测试集则来自 CASP15 基准集中的 6 个天然 RNA 和 4 条人工合成 RNA。
对于 CASP16,实验所用整套训练集由 994 个实验解析的 PDB 结构与 2,170 个用于交叉蒸馏数据增强的高置信度预测结构构成,总计 3,164 个样本。每个训练批次 PDB 结构与交叉蒸馏数据的数量比约为 1:2.2。
具体来看,首先同样道理,CASP16 首批待测 RNA 发布于 2024 年 5 月,因此研究人员将 RNA3DB 中所有符合条件的数据合并成一整套训练集,仅包含 2024 年 4 月 6 日及之前上传的 PDB 结构。经严格筛选处理后,最终得到 994 条 RNA 序列及其对应的实测三维结构。测试数据来自 CASP16 当前可获取的 28 个有真实实验三维结构的 RNA。
构建交叉蒸馏数据集的数据则来自 bpRNA-1m(90) 数据集,主要用于探究数据增强带来的效果。bpRNA-1m(90) 共包含 28,370 条 RNA 序列(及对应的二级结构),在经过去冗余处理并限制序列长度为 30-200,并多次借助 MMseqs2 聚类工具提取后,最终得到了 2,170 个高置信度 RNA 结构的交叉蒸馏数据集。
基于流匹配的条件生成框架
RNAbpFlow 是一款基于序列和碱基对条件的 SE(3) 等变流匹配模型(如下图所示),其基于蛋白质结构生成模型 FrameFlow 改造而来,采用 NuFold 提出的核苷酸表征方式,将 RNA 序列中每个核苷酸表征为刚体标架,以核苷酸序列与碱基配对信息为约束,通过流匹配迭代采样,预测二面角分步还原全部原子,最终完整输出带真实核糖折叠构象的 RNA 全原子三维结构。简而言之,RNAbpFlow 的核心优势可以概括为两点,即「输入条件」和「模型范式」。
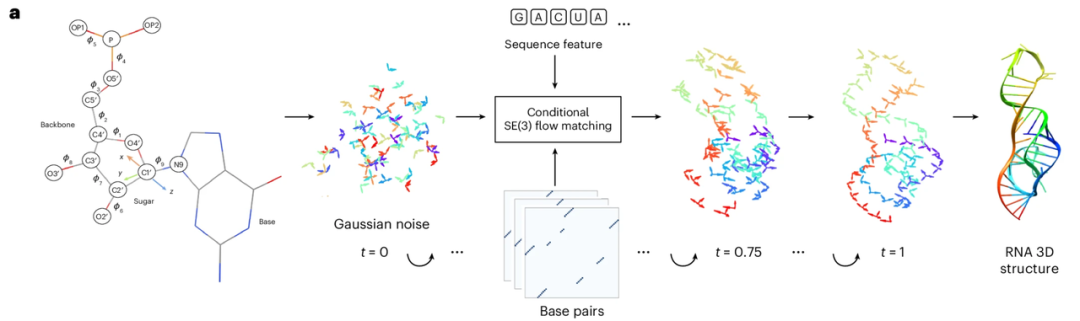
*RNAbpFlow 整体流程图*
就「输入条件」来看,RNAbpFlow 无需任何 MSA 或同源模板信息,仅依靠序列和碱基配对信息即可条件式生成 RNA 三维结构。
首先针对序列,输入一段长度为 L 的 RNA 序列,用独热编码(one-hot encoding)对核苷酸进行表征,即用含 4 个元素的二元向量分别对应四种核苷酸(A、U、C 和 G);其次针对训练阶段的碱基配对信息处理(此为关键创新),借助 RNAView、MC-Annotate 和 DSSR 三款工具,从实验解析的天然三维结构中提取二维配对标注,并将其分别表示为三张独立的 L × L 二元矩阵。为完整捕获各类经典与非经典碱基配对特征,研究不统一修正三款工具各自得到的互相矛盾的标注,而是直接将三张二元矩阵拼接为 L × L × 3 的张量,作为去噪网络结构中偏置项的输入,分三个独立通道提供配对特征信息。
另外针对 CASP15 和 CASP16 竞赛测试靶点的采样推理场景,由于无天然碱基配对真值,实验采用三款 RNA 二维结构预测工具输出的序列依赖型配对预测矩阵,分别是 IPKnot、SPOT-RNA 和 RibonanzaNet。这三款工具均支持假结识别的碱基配对预测,选型依据为其在 CASP15 天然 RNA 靶点上的采样效果。除此外,RNAbpFlow 还具备高度通用性,支持用户自定义三组配对矩阵作为输入,若仅有一组自定义配对矩阵,也可将其复制三份,以匹配网络所需的三通道二维输入格式。
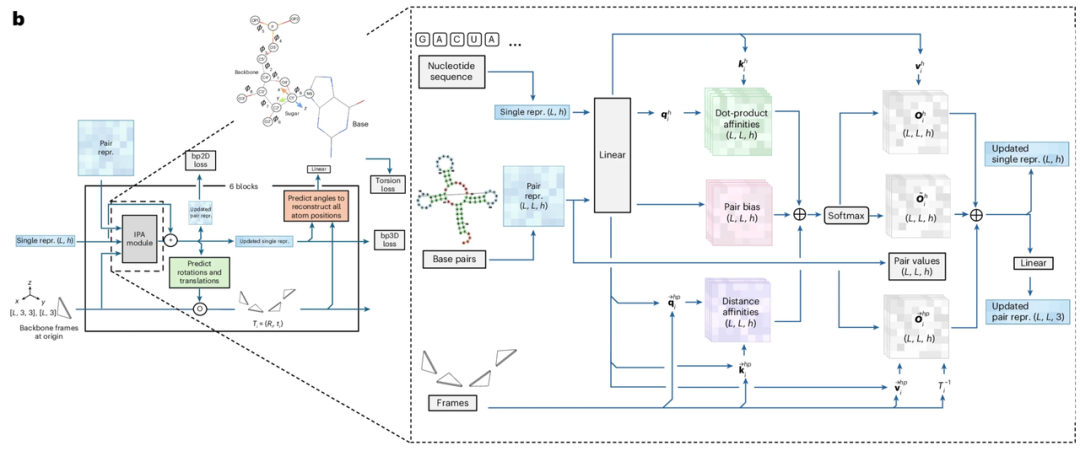
*RNAbpFlow 网络架构图*
就「模型范式」来看,其关键在于「流匹配」。流匹配是一类深度生成模型,其核心目标是学习一个速度场(或称流场),该速度场能够匹配数据分布的概率流,从而将高斯分布这类简单分布转化为高维空间中目标所需的复杂数据分布。流匹配会直接学习该速度场,用以描述样本点从简单分布迁移至目标分布的运动规律,且过程不会彻底破坏原有数据分布。通过学习得到的向量场上积分常微分方程,流匹配能够生成更简洁的迁移轨迹以逼近目标。相比扩散模型等,流匹配可大幅提升大规模样本生成的计算速度。
在本研究中,流匹配方法的目标是学习参数化向量场 Ut。该向量场表示一种光滑、随时间 t 变化的映射,由此生成常微分方程,用于描述两种分布间的转换关系,分别是分布 p₀(T₀)(含噪声帧)与分布 p₁(T₁)(真实基准帧)。为了学习该映射,研究人员训练参数化神经网络 Vθ(Tₜ,t),该网络可根据时间 t 下受噪声污染的真实基准帧 Tₜ 预测向量场。该部分网络参考 FrameFlow 的设计,采用 AlphaFold 2 中的结构模块作为骨干网络架构。
具体训练设置方面,实验基于 PyTorch-Lightning 框架训练模型,采用 Adam 优化器,学习率设置为 0.0001.分布式训练流程在 8 张 NVIDIA H100 GPU 上运行,迭代次数为 1500 轮。
关键任务表现赶超 AlphaFold 3
为了评估 RNAbpFlow 的采样性能,研究首先将其与 RNAJP 进行了对比。后者是一种基于粗粒度分子动力学模拟的 RNA 三维结构采样方法,可显式考量碱基配对信息,涵盖非经典碱基配对、碱基堆积相互作用以及长程环-环相互作用。
实验结果显示(如下图所示),RNAbpFlow 在两项评价指标上的表现均优于 RNAJP。具体来说,RNAbpFlow 的平均局部距离差异测试分数(lDDT score)高达 0.66,而 RNAJP 的表现仅为 0.59;同样,全局拓扑结构采样方面,RNAbpFlow 平均模板建模分数(TM-score)为 0.38,而 RNAJP 为 0.32。
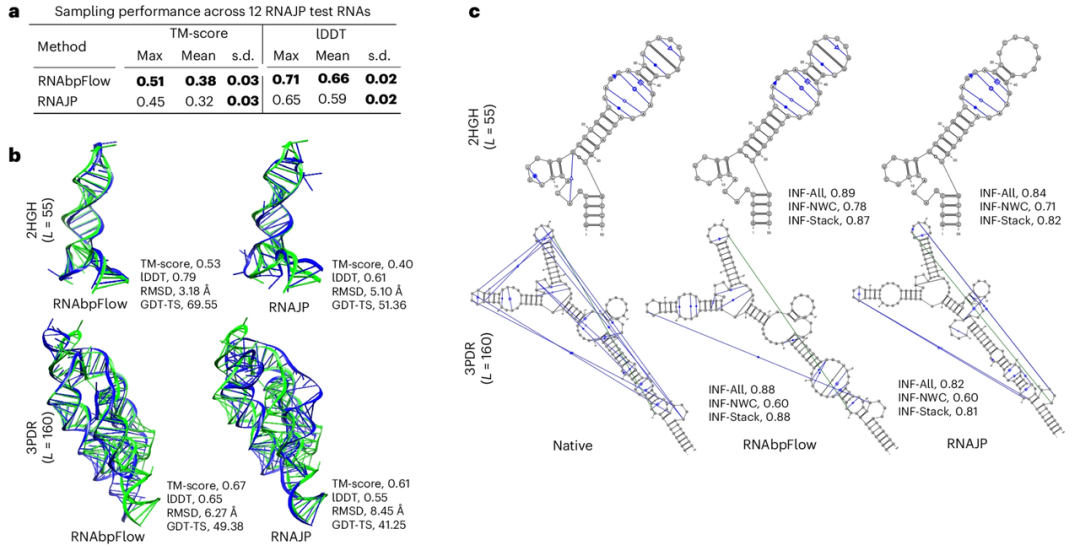
*RNAbpFlow 与 RNAJP 的实验对比*
在采样有效性评估方面,基于 TM-score 判定的正确折叠结构,RNAbpFlow 检出 RNA 靶标的比例高达 66.67%,基于 lDDT 分数判定的正确折叠结构检出占比为 25%;反观 RNAJP 两项占比分别仅为 41.67% 和 0%。另外在 RNAbpFlow 生成的全部 12,000 个模拟构象中,13.4% 的构象模板建模分数高于 0.45,9.6% 的构象 lDDT 分数高于 0.7。与之形成对比的是,RNAJP 的模拟构象中仅有 1.73% 模板建模分数超过 0.45,无任何构象的 lDDT 分数高于 0.75。
以上结果证明,RNAbpFlow 不仅最优结构的打分表现优于 RNAJP,高质量模拟构象的占比也更高,凸显了其在全局拓扑与局部构象双重采样任务中的高效性。
接着,研究团队在 CASP15 盲测数据集上,将 RNA bpFlow 与多种现存方法进行了对比(如下表所示)。结果显示,当输入真实准确的天然碱基配对信息时,RNAbpFlow 的建模预测性能得到显著提升,平均 TM-score 达到 0.48,全原子均方根偏差(RMSD)为 7.77,非沃森-克里克碱基对相互作用网络保真度(INF-NWC)为 0.62;当仅使用算法预测得到的碱基配对时,三项指标分别为 0.40、10.70 和 0.48。
相较之下可见,当提供真实碱基配对信息时,TM-score 提升了 20%、RMSD 下降了 27.4%、INF-NWC 提升了 29.2%,这凸显了高质量碱基配对信息的重要性。
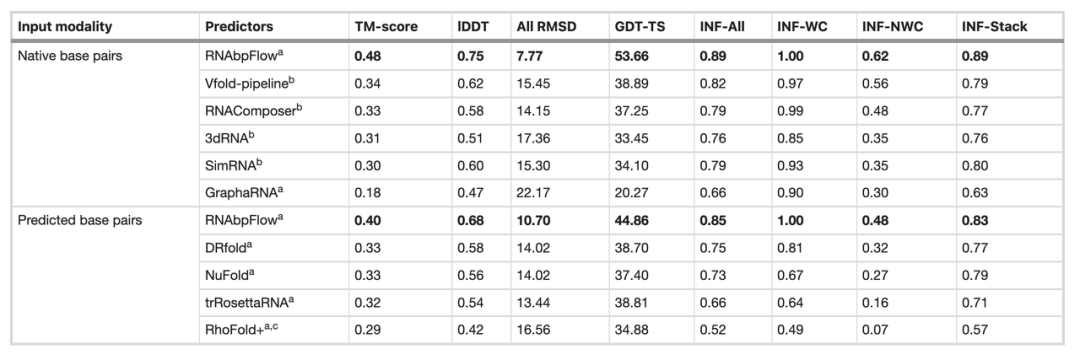
*在 CASP15 盲测中 6 种方法对比结果*
与之形成对比的是,其余方法即便输入真实的天然碱基配对,性能提升的幅度也十分有限,比如剩余方法中最高的 Vfold,其 TM-score 最高也仅为 0.34,RNAComposer 最低 RMSD 为 14.15。这都证明了 RNAbpFlow 具备更强的适配性,能够在深度生成建模过程中高效利用精准碱基配对约束,极大提升了人工智能方法预测性能的上限。
为验证 RNAbpFlow 在 CASP16 盲测靶标上的采样精度,研究人员将其与 CASP16 竞赛中表现最优的两款预测模型进行了对比。结果显示(如下图所示),在 14 个预测目标(≤200nt )的平均最大 TM-score 与 lDDT 指标上,RNAbpFlow 的性能优于包括 AlphaFold 3 在内的所有算法,RNAbpFlow 生成的结构构象集合中 12 个(占比 85.71%)可以找到至少一个正确折叠构象;相比之下,AlphaFold 3 仅有 8 个(57.13%)实现此目标,体现了 RNAbpFlow 所具备更稳定的性能。
不过对于长于 200 nt 的 RNA 靶标,RNAbpFlow 性能虽仍优于 优于 NuFold、trRosettaRNA2 和 DRfold2,但表现却弱于 AlphaFold3,其根源是长序列的预测碱基配对保真度大幅下降。
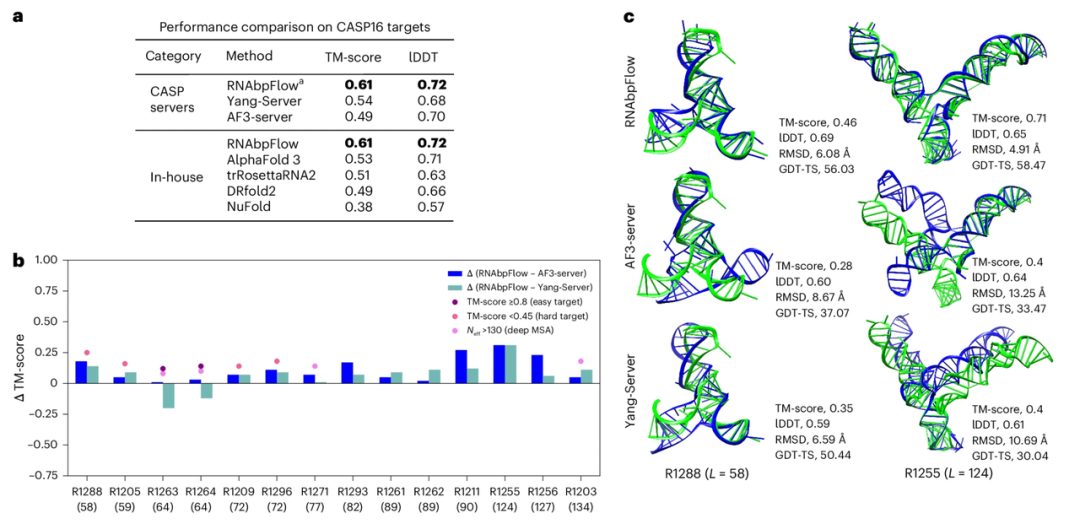
*RNAbpFlow 与包括 AlphaFold 3 在内的先进方法的比较*
此外实验还证明了对于难以预测的目标(TM-score < 0.45;MSA 有效同源序列深度 ≦ 130、进化信号微弱的情况下),在进化信息匮乏时,基于碱基配对约束的结构预测模型显然具备更佳的效果,显示了仅依靠碱基配对信息即可完成 RNA 三维结构预测的 RNAbpFlow 的优越性;而当面对 R1263、R1264 这类容易预测的目标时,充足的深度多序列比对数据使得两款对比模型 AF3-server、Yang-Server 可与 RNAbpFlow 持平甚至更优,体现出两款模型对于对序列对比信息的强依赖性。
写在最后
总的来说,RNAbpFlow 不受 MSA 和结构同源性的限制,是仅靠序列和碱基配对即可端到端直接生成全原子 RNA 三维结构模型,其借助高精度原子级大规模构象集生成技术,或将为 RNA 构象动力学研究开辟一条极具前景的探索新方向。
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢