
作者:文龙 来源:scienceAI
预测药物特性对加速药物发现有着重要的推动作用,基于图的神经网络进行分子表示学习也已经取得了不错的成绩。尽管如此,人类专家对分子表示和卷积神经网络知识的综合潜力尚未得到充分的探索。
近日,清华大学、复旦大学和浙江大学的研究人员联合新加坡国立大学的研究人员开发了一种开箱即用的AI工具,可以通过分析基于人类知识的分子表示来预测药物特性。 他们的研究成果于3月1日以「通过广泛学习的基于知识的分子表示对药物特性进行开箱即用的深度学习预测」(Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations)发表在《自然·机器智能》(Nature Machine Intelligence)杂志上。论文的主要作者是清华的博士生Wan Xiang Shen。
适当的分子表示对于增强网络的学习和预测能力至关重要。现在,主要的分子表示学习方法大致可分为四类:
- 第一种是基于图的特征表示,使用图卷积网络(GCNs)或图注意力网络(GATs)直接从分子的底层图进行从头学习,获取在制药相关任务中的SOTA性能。
- 第二种是字符串表示,使用CNN或RNN对化学结构的字符串表示进行学习,例如SMILES(用ASCII字符串明确描述分子结构的规范)。
- 第三种是图像表示,使用CNN对按规则渲染的2D化学图或Kekulé图进行学习。
- 第四种是基于知识的表示,深度学习模型用于先验的人类知识衍生的分子表示或指纹特征的学习。
该团队对第三种和第四种方法进行了探索,基于CNN架构提出了新的架构——MolMapNet,该架构可以将分子表示和指纹特征映射到健壮的二维特征图,再对这些特征图进行学习,实现对药物特性的预测。
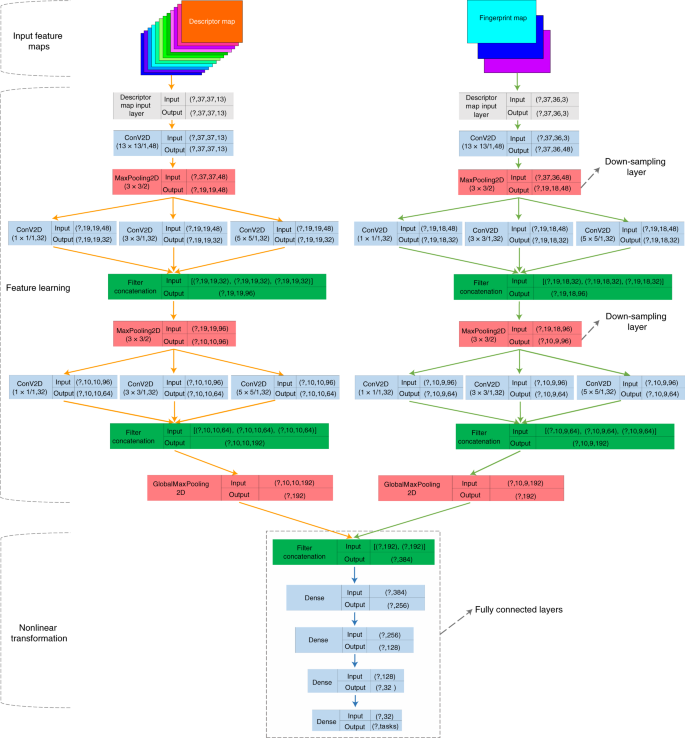
通过与基于图的神经网络方法的对比,实验结果显示MolMapNet模型在26个药学相关的基准数据集和一个新颖的数据集中的表现均具有一定的竞争力,并且由于结合了人类的先验知识,模型更具广泛性。
论文的作者之一,新加坡国立大学计算生物学教授Yu Zong Chen表示:「我们的研究有两个显著成就:首先是引入了一种新方法,用于将无序分子特性映射到表示分子特性的内在关系的有序图像中;其二是开发了一种创新的开箱即用的AI工具,使用具有SOTA性能的非专家深度学习预测药物特性。」
跟多详情可以戳原文。
论文链接:http://dx.doi.org/10.1038/s42256-021-00301-6
项目地址:https://github.com/shenwanxiang/bidd-molmap
参考内容:
https://techxplore.com/news/2021-03-molmapnet-out-of-the-box-deep-pharmaceutical-properties.html
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢