
【论文标题】Multimodal Graph-based Transformer Framework for Biomedical Relation Extraction
【作者团队】Sriram Pingali, Shweta Yadav, Pratik Dutta, Sriparna Saha
【发表时间】2021/07/01
【机 构】印度理工学院巴特那校区
【论文链接】https://arxiv.org/pdf/2107.00596v1.pdf
最近,预训练的Transformer模型的进步推动了各种生物医学任务中有效文本挖掘模型的发展。然而,这些模型主要是在文本数据上学习的,往往缺乏实体的领域知识来捕捉句子以外的背景。在这项研究中,我们引入了一个新的框架,使模型能够在额外的多模态线索(如分子结构)的帮助下学习关于实体(蛋白质)的多模态生物信息。为此,我们没有开发特定模态的架构,而是设计了一个通用和优化的基于图的多模态学习机制,利用GraphBERT模型来编码文本和分子结构信息,并利用各种模态的基本特征来实现端到端的学习。我们在生物医学语料库中的蛋白质相互作用任务上评估了我们提出的方法,其中我们提出的通用方法被观察到受益于额外的特定领域模态。
- 除了生物医学语料库的文本信息外,我们在识别蛋白质相互作用时还利用了蛋白质的原子结构信息。
- 本文开发了一种通用的模态无关的方法,能够学习文本和蛋白质结构模态的特征表示。
- 我们的分析显示,蛋白质结构模态的加入提高了模型在识别相互作用的蛋白质方面的效率
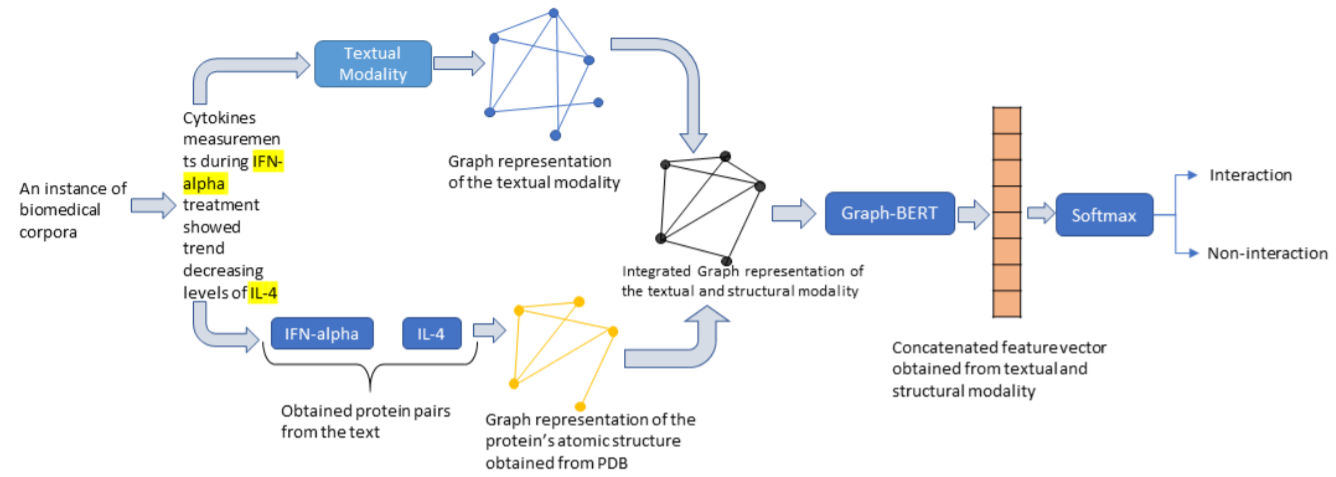
上图为基于生物医学文本的深度多模态PPI预测的框架。
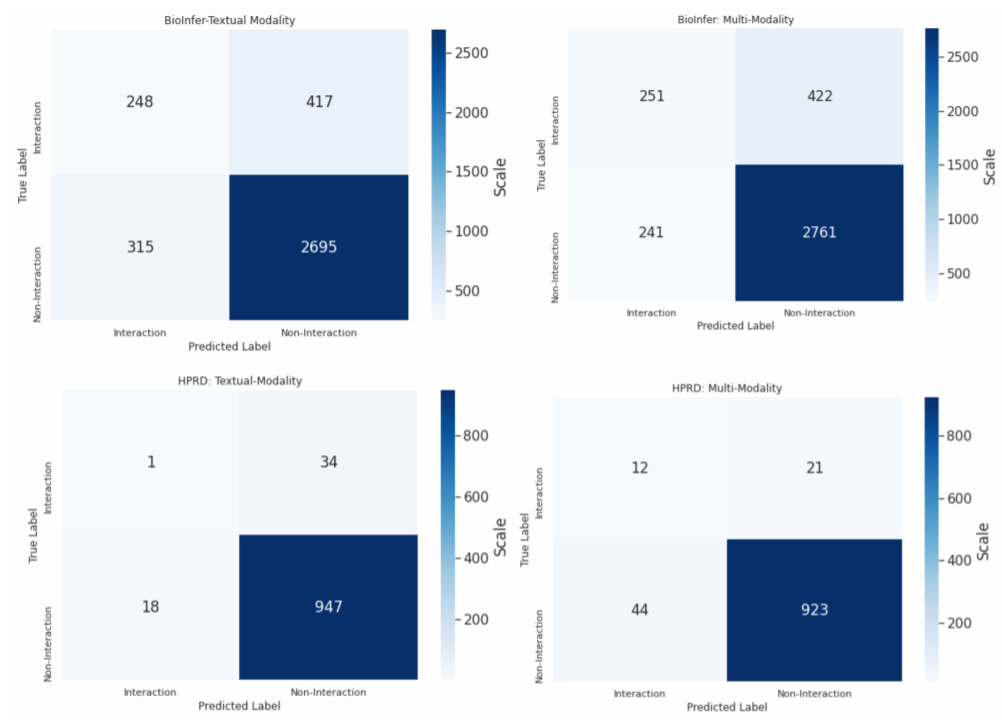
在2个PPI预测标准任务,bioInfer和HPRD50上分别在多模态和单模态下得到的混淆矩阵的结果。文章中得到了以下结论:
- 在一个句子中提到大量的蛋白质的实例会导致错误分类。例如,BioInfer和HPRD50数据集的任何实例中,蛋白质的最大数量分别为26和24。这些存在于单个实例中的大量蛋白质可能导致网络的错误分类。
- 少数样本包含同一蛋白质的重复提及。这增加了噪音,并可能导致失去有用的背景信息。
- 为了从分子结构中得到一个一致的图,要求节点的长度相同。这是通过用零填充向量来实现的,当PDB不可用时,为了保持一致性,会使用一个空向量。更好地处理缺失数据将有助于学习提议的模型。
这项工作提出了一个新的模式无关的基于图的框架来识别蛋白质之间的相互作用。具体来说,我们探索了两种模式:文本和分子结构,使模型能够学习特定领域的多分子信息与特定任务的背景信息相辅相成。详细的比较结果和分析证明,我们提出的多模态方法可以捕获潜在的分子结构信息,而无需依赖复杂的特定模态架构。未来的工作旨在将这项研究扩展到其他相关的任务,如药物-药物相互作用。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢