
【论文标题】Single-sequence protein structure prediction using language models from deep learning
【作者团队】 Ratul Chowdhury, Nazim Bouatta, Surojit Biswas, Charlotte Rochereau, George M Church, Peter Karl Sorger, Mohammed N AlQuraishi
【发表时间】2021/08/04
【机 构】哈佛大学医学院、哥大
【论文链接】https://www.biorxiv.org/content/10.1101/2021.08.02.454840v1.full.pdf
【推荐理由】单序列蛋白质结构预测与AlphaFold2和RoseTTAFold的比较
AlphaFold2和相关系统使用深度学习,从多重序列比对(MSA)中编码的共进化关系中预测蛋白质结构。尽管最近准确率大幅提高,但仍有三个挑战,预测无法生成MSA的孤儿和快速进化的蛋白质,快速探索设计的结构,以及了解溶液中自发多肽折叠的规则。在这里,我们报告了一个端到端的可微的递归几何网络(RGN),它能够在不使用MSA的情况下从单个蛋白质序列预测蛋白质结构。这个深度学习系统有两个新的元素:一个是蛋白质语言模型AminoBERT,它使用Transformer从数以百万计的未比对的蛋白质中学习潜在的结构信息;另一个是几何模块,它紧凑地表示Cα骨架几何。RGN2在孤儿蛋白上的表现优于AlphaFold2和RoseTTAFold(以及trRosetta),并在设计序列上具有竞争力,同时在计算时间上实现了高达10万倍的减少。这些发现证明了蛋白质语言模型相对于MSA在结构预测方面的实际和理论优势。
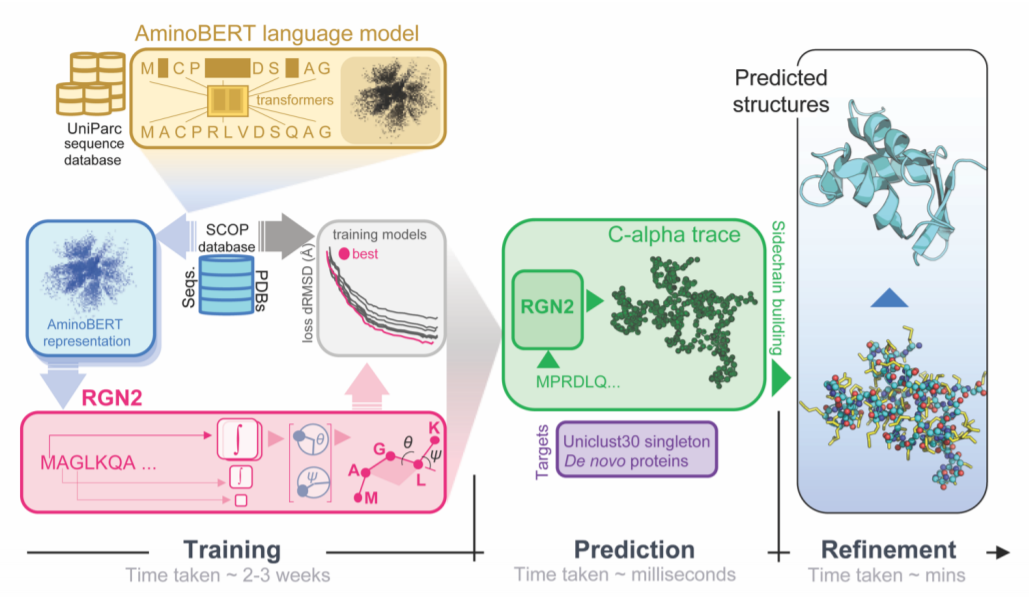
上图展示了RGN2的框架和工作流。RGN2结合了基于Transformer的蛋白质语言模型(AminoBERT)和循环几何网络,利用Frenet-Serret框架来生成蛋白质的骨架结构,随后使用Rosetta能量函数进行侧链原子的放置和氢键网络的细化。
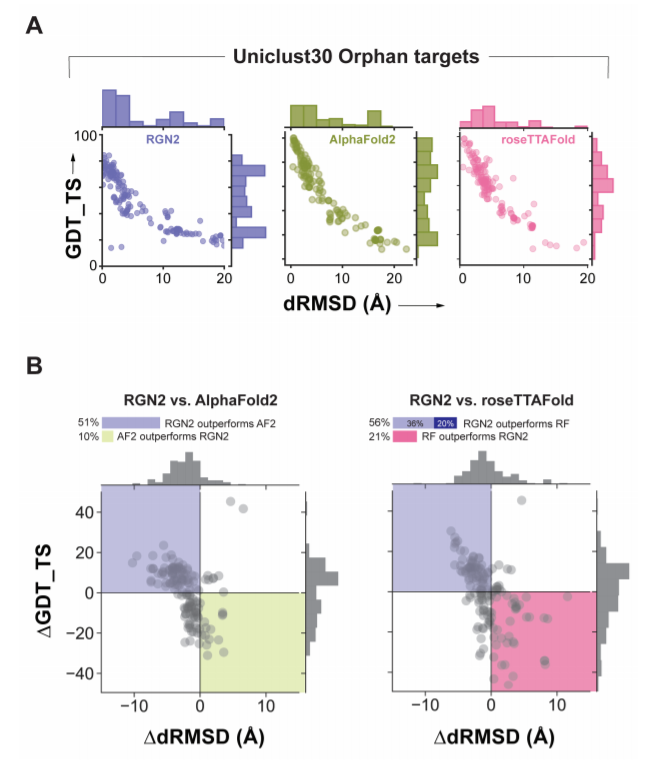
A图:RGN2(紫色)、AF2(绿色)和RF(粉色)在196个缺乏已知同源物的孤儿蛋白中的性能指标。RGN2图中的离群值对应于短靶点,其中几个预测不佳的残基大大降低了GDT_TS。
B图:使用dRMSD和GDT_TS作为指标,显示了196个孤儿蛋白的RGN2和AF2/RF之间预测准确性的差异。RF对40个目标未能收敛,没有产生预测结果。左上角的点对应于ΔdRMSD为负、ΔGDT_TS为正的目标,即RGN2在这两个指标上都优于竞争对手的方法。在右下角象限反之亦然。其他两个象限(白色)表示在两个指标不一致的情况下没有明显的赢家。
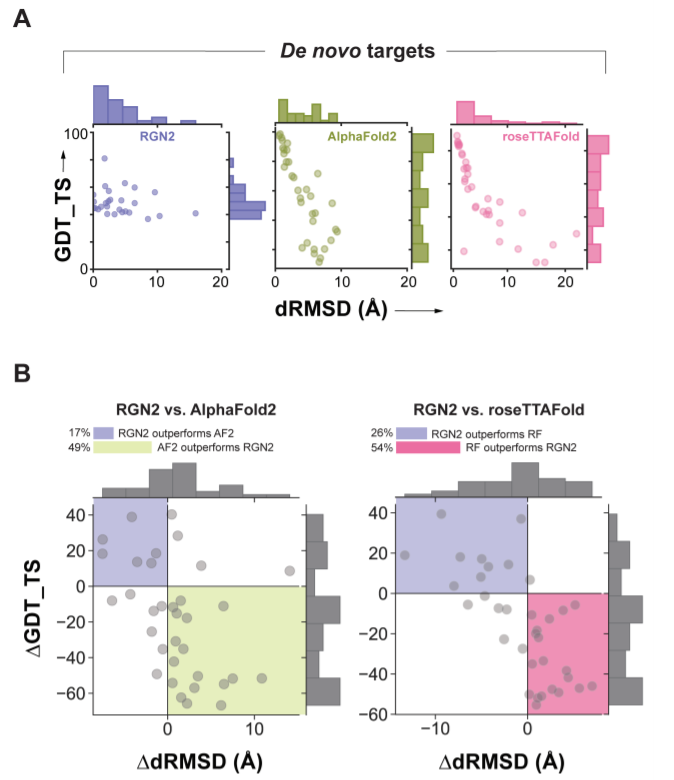
上图展示了三种方法,RGN2(紫色)、AF2(绿色)和RF(粉色)在35个没有已知同源物的新设计蛋白质中的性能指标,以及使用dRMSD和GDT_TS作为指标,显示了这35个蛋白质的RGN2和AF2/RF之间的预测准确性差异。我们发现,RGN2分别在17%、26%和45%的情况下在两个指标上都优于AF2、RF和trRosetta,但在49%、54%和7%的情况下在两个指标上表现不佳。
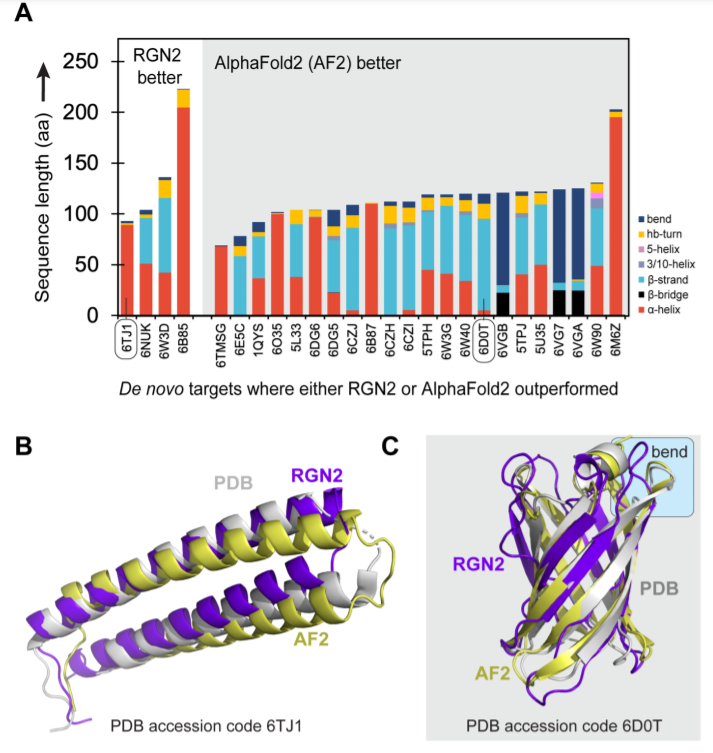
A图:堆积的柱状图显示了35个新蛋白质中二级结构的相对比例,高度表示蛋白质的长度,AF2在具有高β-片层含量的蛋白质上表现优于RGN2。
B图:α螺旋靶点6TJ1没有弯曲,但AF2预测了一个虚假的弯曲,而RGN2的预测则更接近实验结构。
C图:6D0T,一个由无序环连接的 hour-glass 形状β-barrel的组合,被AF2预测的更好。
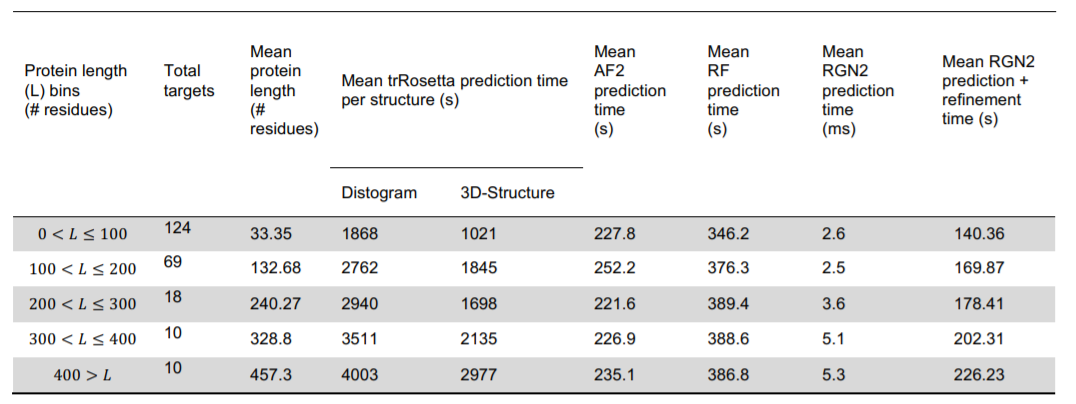
上图展示了RGN2与AF2、RF和trRosetta在231个目标上的预测时间比较,这些靶点包括了孤儿和de novo的蛋白质数据集。RGN2的预测是分批进行的,最大允许的批次大小为128个目标。由于没有一个目标有已知的同源蛋白,所以没有使用trRosetta MSA生成步骤。
- 结论
本文的结论是,RGN2可以为蛋白质空间的新区域学习序列结构关系,但从单一序列预测β片仍然是一个挑战。相对于AF2而言,RGN2在non-trivial的新蛋白质上的优异表现也表明,这两种方法可能是互补的,混合模型的表现可能优于单独的一种。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢