
【论文标题】Learning interpretable cellular and gene signature embeddings from single-cell transcriptomic data
【作者团队】Yifan Zhao, Huiyu Cai, Zuobai Zhang, Jian Tang & Yue Li
【发表时间】2021/09/06
【机 构】Mila、麦吉尔大学等
【论文链接】https://www.nature.com/articles/s41467-021-25534-2
【代码链接】https://www.github.com/hui2000ji/scETM
单细胞RNA测序(scRNA-seq)技术的出现彻底改变了转录组研究。然而,scRNA-seq数据的大规模综合分析仍然是一个挑战,主要原因在于批次差异和现有计算方法的可迁移性、可解释性和可扩展性有限。作者提出了单细胞嵌入式主题模型(scETM)。作者的主要贡献是利用了一个可迁移的基于神经网络的编码器,同时通过矩阵处理拥有一个可解释的线性解码器。特别是,scETM同时学习一个编码器网络来推断细胞类型混合物和一组高度可解释的基因嵌入、主题嵌入等。利用基因组富集分析,作者发现scETM学习的主题富集在有生物学意义的和与疾病相关的途径中。最后,scETM能够将已知的基因集纳入基因嵌入,从而通过主题嵌入直接学习路径和主题之间的关联。
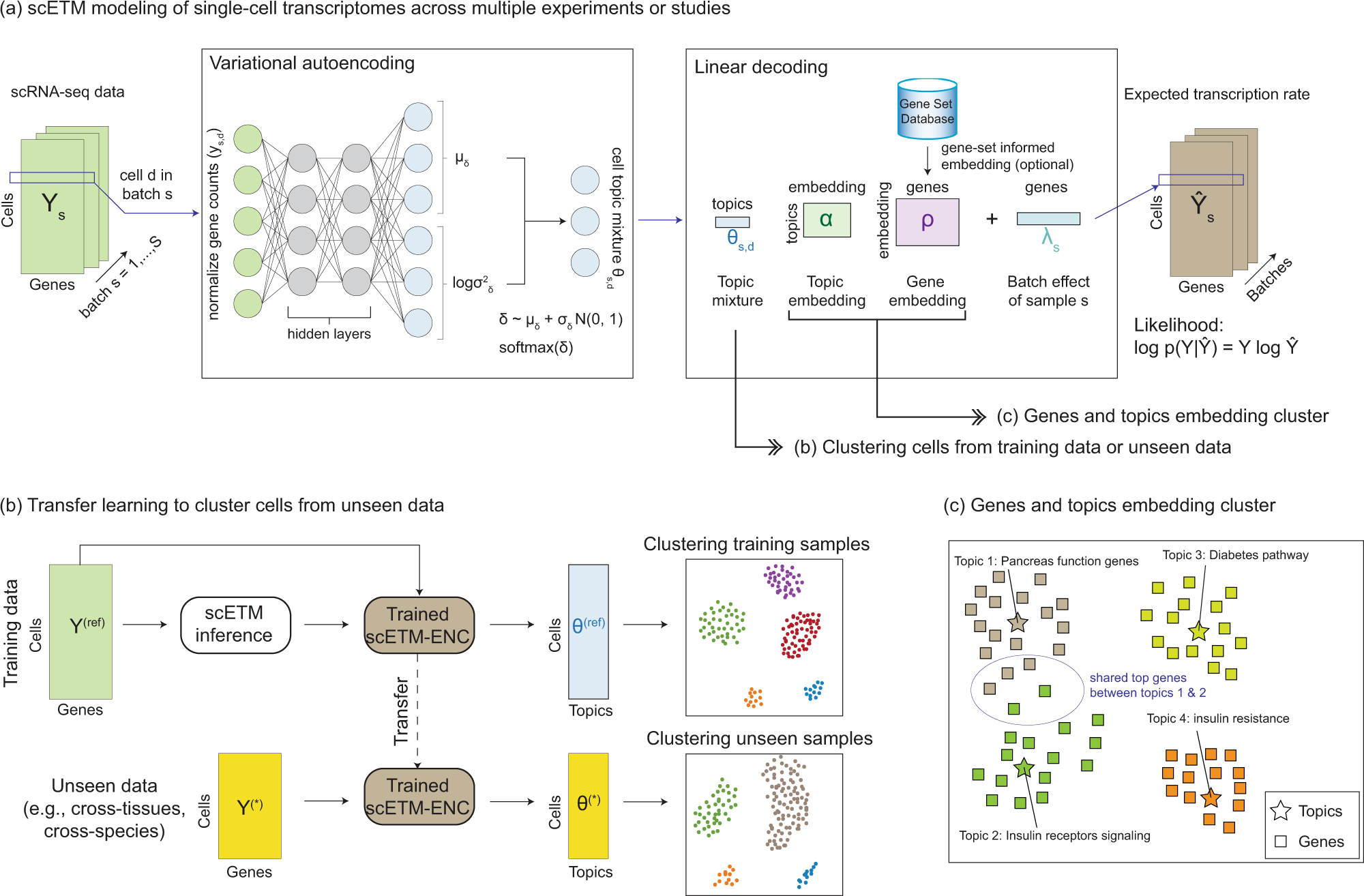
a图:scETM训练。鉴于跨多个实验或研究(即批次)的scRNA-seq数据矩阵作为输入,scETM使用嵌入式主题建模方法对单细胞转录组进行建模。每个scRNA-seq谱作为归一化基因计数输入到变异自动编码器(VAE)。编码器网络产生潜在主题混合物的随机样本(θs,d为批次s=1, ..., S和细胞d=1, ..., Ns),可用于细胞聚类。线性解码器学习主题嵌入和基因嵌入,可用于通过富集分析来分析细胞程序。
b图:用来进行零次迁移学习的工作流程。在参考scRNA-seq数据集上训练好的scETM-编码器被用来推断未见过的scRNA-seq数据集的细胞主题混合物θ*,而不需要训练它们。然后通过UMAP可视化产生的细胞混合物,并通过标准的无监督聚类指标使用地面真实的细胞类型进行评估。
c图:探讨基因嵌入和主题嵌入。由于基因和主题共享相同的嵌入空间,作者可以通过UMAP可视化来探索它们的联系,或者通过使用已知路径的富集分析来注释每个主题。
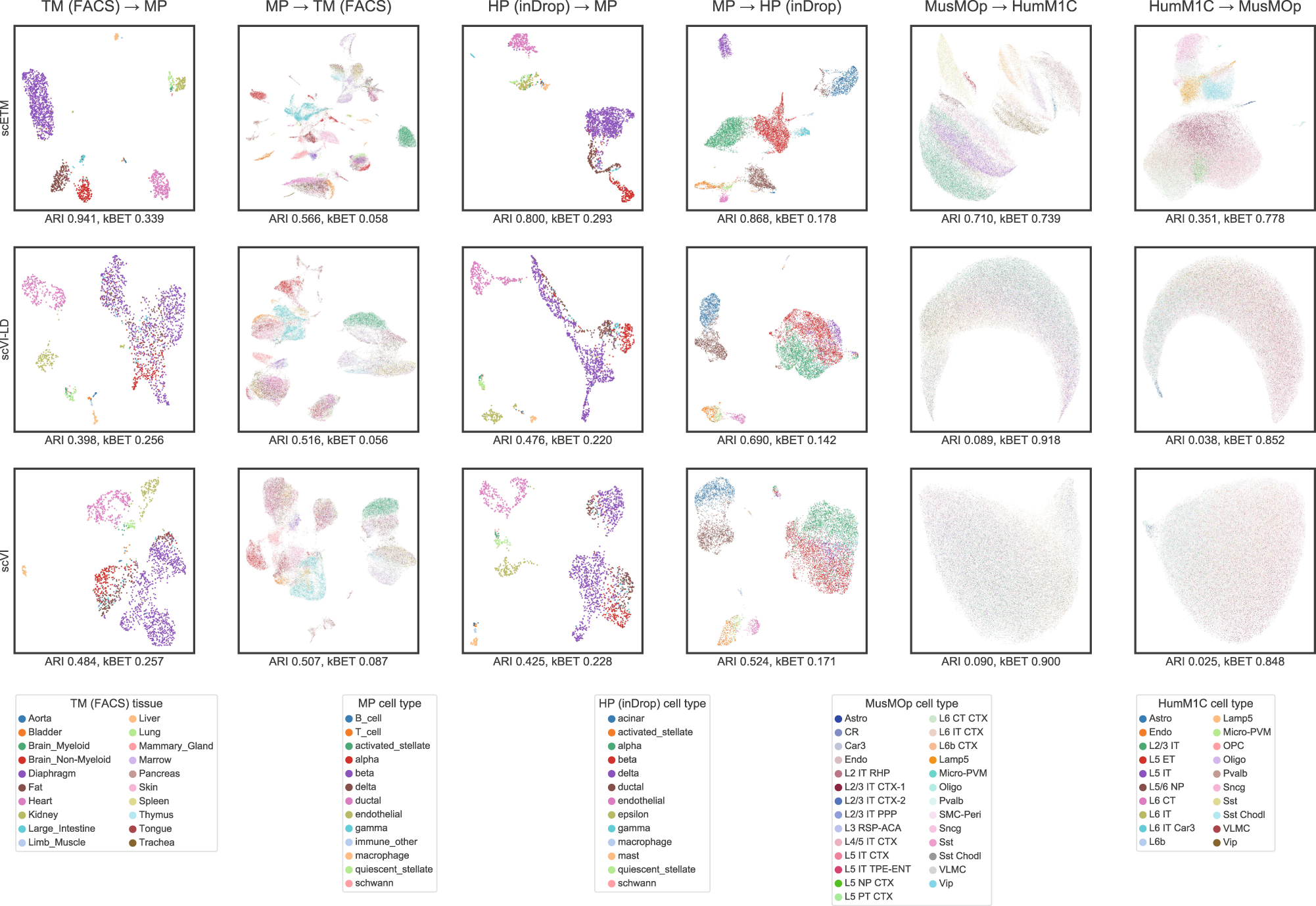
每个面板都显示了通过在数据集A上训练和应用于数据集B(即A→B)的细胞的UMAP可视化。作者总共进行了6次迁移学习任务。TM(FACS) ↔ MP,HP(inDrop) ↔ MP,MusMOp ↔ HumM1C。对于每个任务,作者评估了scETM、scVI-LD或scVI,如图所示,细胞是按组织或细胞类型着色的。相应的ARI(Adjusted Rand Index )和kBET(k-nearest-neighbor Batch-Effect Test)在每个板块下面标明。
注释:TM(FACS):用荧光激活单细胞分选法对tabula muris进行测序;MP:小鼠胰腺;HP(inDrop):用InDrop技术对人类胰腺进行测序;HumM1C:人类初级运动皮层(来自艾伦大脑地图);MusMOp:小鼠初级运动区(来自艾伦大脑地图)。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢